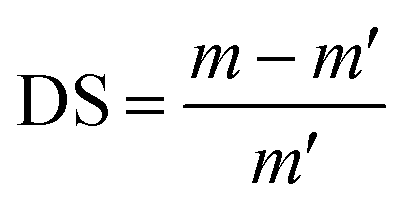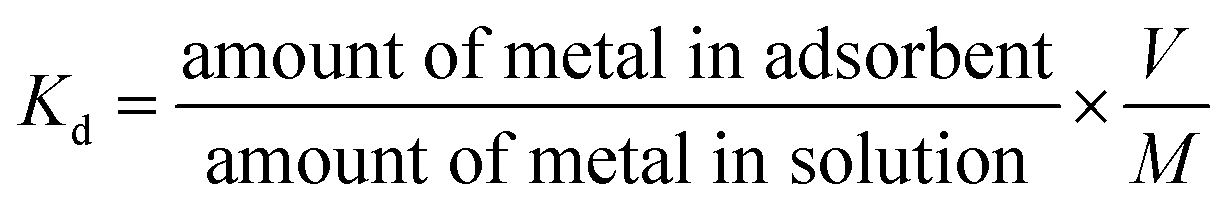DOI:
10.1039/C5RA10264B
(Paper)
RSC Adv., 2015,
5, 74220-74229
Uranium(VI) adsorption from aqueous solutions using poly(vinyl alcohol)/carbon nanotube composites
Received
4th June 2015
, Accepted 24th August 2015
First published on 24th August 2015
Abstract
Poly(vinyl alcohol)/multiwalled carbon nanotubes (PVA/MWCNTs) composite hydrogels were prepared by a dispersion method and their ability to adsorb and remove uranyl ions from aqueous solutions was investigated. The prepared composites were characterized by XRD, TEM, SEM and FTIR. The effect of contact time, solution pH, initial UO22+ ion concentration and temperature on UO22+ ion adsorption from aqueous solution onto the prepared hydrogels was studied. The obtained results illustrated that, dispersion of MWCNTs into the PVA matrix enhanced the removal efficiency of UO22+ ions compared to PVA only. The Langmuir and Freundlich adsorption models have been applied to evaluate the adsorption efficiency and the data correlated well with the Langmuir model. Thermodynamic parameters (ΔH°, ΔS°, ΔG°) were determined which indicated that the UO22+ ion adsorption process onto the prepared hydrogels was exothermic and spontaneous. The adsorbed UO22+ can be desorbed effectively by 0.1 M EDTA.
1. Introduction
Uranium is a radioactive element widely distributed over the earth's crust and it has a significant importance in nuclear industry since it is used as a fuel for nuclear power plants. In general, uranium released into the environment is often dissolved in aqueous solutions in the hexavalent form as UO22+. Due to its strong radiation, uranium contamination can cause serious environmental problems;1 therefore, uranium removal from aqueous solutions is important in view of nuclear fuel resources and human health.2 In the last decades several techniques were developed for UO22+ ions removal from aqueous solutions. These techniques include precipitation, co-precipitation, solvent extraction, membrane dialysis, chromatographic extraction, ion exchange, floatation and adsorption.3–8 Among these techniques adsorption seems to be the most attractive one due to the advantages it has such as low cost, ease of operation, wide availability of adsorption materials and high resistance towards toxic chemical compounds in water.8 Various kinds of new adsorbents for uranium removal and recovery have been reported,9–11 however, the adsorption capacity and selectivity of those adsorbents towards UO22+ ions need to be improved. Hydrophilic matrix adsorbents e.g. hydrogels as well as polymeric composites were also widely used to enhance the adsorption capacity of uranyl ions from aqueous solutions.12,13
Nano-sized materials have attracted substantial interest in the scientific communities due to their unique physical and chemical properties such as large surface-to-volume ratio, high bioactivity, excellent conformation stability, good biocompatibility, excellent conductivity and catalytic efficiency.14 Furthermore, nanoparticles surface atoms are unsaturated, and can, therefore, bind with other atoms that feature high chemical activity. In recent years, various kind of nano-sized materials have been prepared15 and found potential applications in many fields such as photocatalysis,16 optical sensors17 and medical applications.18 Nanomaterials including traditional inorganic nanoadsorbents and novel polymer supported composites have been extensively applied for pollutants removal from aqueous solutions, due to their novel size- and shape-dependent properties, contributing to their excellent removal efficiency.19,20
Multiwalled carbon nanotubes (MWCNTs) have inherent extraordinary structural, mechanical, thermal and electronic properties which made them attractive in the field of radionuclide adsorption.21 Polymeric/MWCNTs nanocomposites have been applied extensively in uranium adsorption due to their highly porous and hollow structure, large specific surface area and light mass density.22,23 However, problems associated with the dispersion of MWCNTs fillers and load transfer across the polymer-CNTs interface may limit polymer/CNTs composites usage. The difficulty in preparing well dispersed MWCNTs composite solutions resulted from their high specific surface area which resulted in very strong van der Waals interactions. Therefore, it is of great significance to improve the dispersion of MWCNTs into the polymeric composites. Many efforts have been made to overcome these barriers including using of surfactants,24 high shear mixing,25 chemical modification26 and in situ polymerization.27 Surfactants are widely used to disperse MWCNTs uniformly without breakage and aggregation. It was reported that, using SDS in the preparation of PVA/CNTs composites resulted in good nanotube dispersion and load transfer from polymer matrix to the carbon nanotubes and consequently improve the composites properties.23
Poly(vinyl alcohol) (PVA) is a water-soluble, cheap, nontoxic, hydrophilic, biocompatible synthetic polymer containing large amounts of hydroxyl groups.28 PVA has excellent film forming, emulsifying and adhesive properties, high tensile strength and good flexibility. PVA blending with other polymers and fillers widens its range of applications.29 Further improvement of PVA properties was achieved by synthesis of PVA/MWCNTs nanocomposites.30,31 It was reported that PVA/CNTs composites have a wide range of applications, from biomedical to electromechanical32 and their properties depend on the attributes and dispersion of MWCNTs.33 PVA is a promising candidate in UO22+ ions removal, because of its high degree of swelling in water, high durability and chemical stability.34,35
The purposes of the current study are: (1) to present a simple and environmental friendly method for preparation of PVA/MWCNTs nanocomposites; (2) to improve the dispersion of MWCNTs in PVA matrix through sodium dodecyl sulphate treatment; (3) to characterize the prepared PVA/MWCNTs nanocomposites by X-ray diffraction (XRD), scanning electron microscopy (SEM) and Fourier transform infrared (FTIR) spectroscopy; and (4) to investigate the feasibility of UO22+ ions adsorption by the prepared composites under different experimental conditions.
2. Experimental
2.1. Materials
PVA with average Mw, 127![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 and the degree of hydrolysis 89%, was purchased from Merck, (Germany). Epichlorohydrin (E), potassium hydroxide, sodium dodecyl sulphate (SDS) and other chemicals were obtained from Beijing chemical reagent factory (China) and used without further purification. MWCNTs were supplied from the Egyptian Petroleum Research Institute. According to the product specification, the as-grown MWCNTs have an average particle size of 20–30 nm and purity above 97%. Oxidized MWCNTs were prepared by oxidization with a mixture of concentrated nitric acid and sulfuric acid (1:3, v/v).36 The surface area of MWCNTs was determined using N2-BET method (Micromeritics Surface Area and Porosity Analyzer, ASAP 2020, American) and was found to be 99 m2 g−1. UO22+ stock solution was prepared by dissolving significant amount of [UO2(NO3)2·6H2O], supplied by Mallinckrodt Company, in deionized water.
000 and the degree of hydrolysis 89%, was purchased from Merck, (Germany). Epichlorohydrin (E), potassium hydroxide, sodium dodecyl sulphate (SDS) and other chemicals were obtained from Beijing chemical reagent factory (China) and used without further purification. MWCNTs were supplied from the Egyptian Petroleum Research Institute. According to the product specification, the as-grown MWCNTs have an average particle size of 20–30 nm and purity above 97%. Oxidized MWCNTs were prepared by oxidization with a mixture of concentrated nitric acid and sulfuric acid (1:3, v/v).36 The surface area of MWCNTs was determined using N2-BET method (Micromeritics Surface Area and Porosity Analyzer, ASAP 2020, American) and was found to be 99 m2 g−1. UO22+ stock solution was prepared by dissolving significant amount of [UO2(NO3)2·6H2O], supplied by Mallinckrodt Company, in deionized water.
2.2. Preparation of PVA/CNTs/E and PVA/CNTs/SDS/E composite hydrogels
The synthesis procedure for a typical PVA/MWCNTs nanocomposite (3.0 wt%) was as the following: MWCNTs powder (30 mg) was dispersed in distilled water (15 mL) in an ultrasonic bath (Branson 2510) for 60 min at room temperature. Subsequently, an aqueous solution (10 mL) of PVA (1.0 g) was added to the MWCNTs suspension. Sonication was continued for an extra 60 min to yield a stable black-colored suspension. Afterwards, stoichiometric amounts of the crosslinker (epichlorohydrin) and potassium hydroxide solution were added separately to the suspension with additional sonication for 30 min at room temperature.37 Finally, this homogeneous PVA/CNTs/E mixture was poured into a Teflon Petri dish and kept at 50 °C for film formation until its weight reached an equilibrium value. This previous procedure was repeated but in absence the MWCNTs to produce PVA/E film.
PVA/CNTs/SDS/E composite hydrogel was prepared by mixing MWCNTs powder with SDS solution (30 wt%) (MWCNTs:SDS = 1:10) and sonication gently at room temperature for 5 hours to form a stable aqueous dispersion of MWCNTs which was kept at room temperature for 72 hours, then the abovementioned procedure was repeated to produce PVA/CNTs/SDS/E film.
2.3. Structural analysis
Chemical structures of the prepared hydrogels were analyzed by FTIR system (Shimadzu 8001, Japan). Morphological evaluations of the prepared hydrogels before and after UO22+ ions adsorption were made using SEM (JEOL, 6510 LA, Japan). Transmission electron microscope (TEM) images of the prepared nanocomposites were obtained using TEM (JEOL, JEM-2100, Japan) at an accelerating voltage of 200 kV, after samples ultrasonication in deionized water for 20 min and dispersion on copper grids. XRD (PANalytical, X'pert PRO, Germany) was used to investigate the structure of the prepared nanocomposites. The XRD measurements were carried out in the 2θ angle with the range of 5–70°. Energy-Dispersive X-ray Spectrometer (EDX) (JEOL, 6510 LA) was used to confirm UO22+ ions adsorption by detecting U lines.
2.4. Swelling behavior of the prepared hydrogels
A known weight of the sample disc was immersed in solutions of different pHs (3, 5 and 7) at 30 °C until the swelling equilibrium was reached. The disc was removed, dried with absorbent paper to get rid of excess water then weighed. The degrees of swelling (DS) for the prepared hydrogels were calculated at different time intervals using the following equation:| |
 | (1) |
where m and m′ denote the weights of hydrogel and dried hydrogel sample, respectively.38
2.5. Uranyl ions adsorption studies
Adsorption of UO22+ ions from aqueous solutions onto the prepared hydrogels was investigated in a batch-wise method. Aqueous solutions (50 mL) containing different amounts of UO22+ ions were incubated with the hydrogels (0.03 g) at different initial pHs, (adjusted with 0.1 mol L−1 HNO3 and 0.1 mol L−1 NaOH) and allowed to equilibrate for different conditions while being shaken continuously. Aqueous solutions were separated from the hydrogels at desired intervals and the residual concentrations of UO22+ ions were determined by the Arsenazo-III spectrophotometric method using UV-Visible spectrophotometer (Thermo Evolution 300, England). The amount of UO22+ ions adsorbed per unit mass of the hydrogels was calculated using the following expression:| |
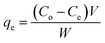 | (2) |
where, qe is the adsorption capacity of the hydrogels (mg g−1); Co and Ce are the concentrations of UO22+ ions in the initial and equilibrium solution (mg L−1), respectively, V is the volume of the aqueous solution (L) and W is the mass of dry hydrogels (g).
To investigate the desorption ability of adsorbed UO22+ from PVA/MWCNTs nanocomposites, desorption experiments were carried using 0.1 M HNO3, H2SO4 and EDTA. After the adsorption reached equilibrium, PVA/MWCNTs composites loaded with UO22+ ions were separated, washed, dried, added to 25 mL of the desired eluent for desorption and shacked at 150 rpm for 4 h. After that the adsorbent was separated washed and dried for the successive adsorption–desorption cycles.
Experimental work using uranyl nitrate hexahydrate was carried out in the safeguards destructive analysis laboratory (KMP-I) at the Egyptian Nuclear and Radiological Regulatory Authority (ENRRA).
3. Results and discussion
3.1. Structural evaluation
FTIR spectra of the prepared MWCNTs powder, PVA/E, PVA/CNTs/E and PVA/CNTs/SDS/E were recorded and shown in Fig. 1. Peaks at 1000–1300 cm−1 are attributed to the absorption of stretching vibration of C–O bonds. The other peaks at 1616 and 1386 cm−1 can be assigned to the nitro groups on the surface of carbon materials, as a result of the efficiency of the modification with nitric acid that was demonstrated by a significant increase in the C–O and –NO functional groups. Peaks at 2921–2930 cm−1 are correlated to C–H stretching. Peaks at 3230–3330 cm−1 are assigned to –OH stretching vibration. The intramolecular and intermolecular hydrogen bonds of the OH groups of PVA and other molecules shifted the band of –OH group to lower frequencies as shown in the PVA spectra. It is noted that there is no characteristic change observed in the peaks around the wavelength range of 3200–3600 cm−1 for PVA/CNTs/E and PVA/CNTs/SDS/E composite hydrogels, suggesting that the dispersion of the incorporated CNTs within the film was essentially physical and the basic chemical characteristic of the prepared material remained intact after their incorporation. Peaks observed at 1221 and 1146 cm−1 of PVA/CNTs/SDS/E spectra are assigned to the sulfate groups of SDS. Peak at 1060 cm−1 in all spectra is for (C–O–C) as a result of formation of crosslinked network structures. The peak around 1095 cm−1 denotes C–O stretching of the secondary alcoholic groups.28,37
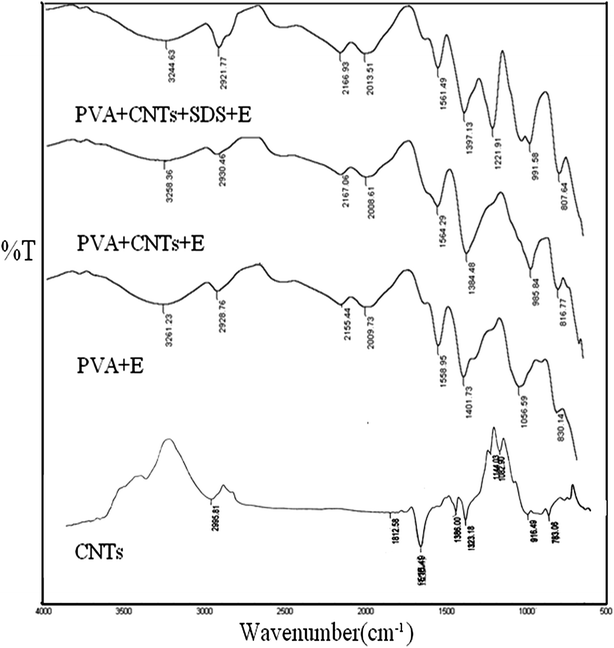 |
| | Fig. 1 FTIR spectra of CNTs, PVA/E, PVA/CNTs/E and PVA/CNTs/SDS/E hydrogel samples. | |
The XRD profiles of MWCNTs powder, PVA/E, PVA/CNTs/E and PVA/CNTs/SDS/E samples were represented in Fig. 2. The characteristic diffraction peak of CNTs appeared clearly at 2θ = 26.2°, implying that its d-spacing resembles the one of pristine graphite. Whereas, the diffraction peaks of PVA/E film are found at 18.7° (main), as well as 9.7, 20.1 and 29.7° (minor).39 The XRD pattern of the PVA/CNTs/E composite film has some peaks which were assigned to CNTs and PVA/E. This means that CNTs were not individually dispersed in PVA matrix but rather in the form of few-layer CNTs. In addition, PVA/E composite film shows an amorphous peak at 19.4°, however CNTs sheets shows a peak of diminished intensity at 23.4°. This result indicates that CNTs had been efficiently exfoliated within the PVA matrix. The crystalline structure of CNTs may be as a result of CNTs overload which is not intercalated into the PVA matrix. On the other hand, PVA/CNTs/SDS/E composite shows the exfoliated peak for PVA composite film and a weak peak for CNTs. This revealed that CNTs were well exfoliated due to its homogenous dispersion in PVA matrix by adding SDS.39
 |
| | Fig. 2 XRD spectra of CNTs, PVA/E, PVA/CNTs/E and PVA/CNTs/SDS/E hydrogel samples. | |
3.2. Water content
The swelling behavior of the prepared hydrogels at different time intervals was investigated and illustrated in Fig. 3. The water affinity of the pure hydrogel (PVA/E) may be attributed to the PVA hydroxyl group, since this hydrophilic group is capable of forming hydrogen bond with water molecules.39 The hydrogel containing SDS relatively swelled in a higher degree compared to PVA/CNTs/E and PVA/E hydrogels. This may be due the fact that, the hydrophobic backbone of SDS interacts with MWCNTs though hydrophobic interaction and thereby anchors the SDS molecules onto the surface of MWCNTs, leaving the hydrophilic head groups to interact with the polymer,40 resulting in increasing the possibility of water uptake into the film and enhancement of the swelling behavior.
 |
| | Fig. 3 The degree of swelling (DS) of PVA/E, PVA/CNTs/E and PVA/CNTs/SDS/E hydrogels at different time intervals (pH = 3). | |
3.3. Microscopic investigations
The surface morphology of the prepared hydrogels, before and after UO22+ ions adsorption, was investigated using SEM and represented in Fig. 4. It is clear from SEM images that there is an obvious change in PVA morphology by introduction of CNTs and the morphology of PVA/CNTs resembles a hybrid composition of its components, while the dark colored textural appearances on PVA after CNTs inclusion are due to the impression of CNTs crystallites. Eventual consequence of IR, XRD and SEM reveals the homogeneous formation of the composites and uniform dispersion of MWCNTs in PVA network. It can be observed from Fig. 4 that, all composites are porous; and have rough surface especially for composites containing CNTs and SDS. It can be also observed that, pores size or pores-content has considerably increased after UO22+ ions adsorption. Uranium adsorption process results in some destruction of the hydrogels ordered structure which may be attributed to the electrostatic repulsion of the partially protonated adsorption sites of the hydrogels with the positively charged UO22+ ions.41
 |
| | Fig. 4 Scanning electron micrographs of: PVA/E, PVA/CNTs/E, PVA/CNTs/SDS/E, hydrogel samples before and after adsorption of UO2 ions. | |
Fig. 5 shows the characteristic TEM image of MWCNTs. This micrograph shows that CNTs have smooth surface and integrated hollow tubular structure and most of the impurity phases, such as amorphous carbon and graphitic nanoparticles, were removed. The diameter of the MWNTs was 20–30 nm.
 |
| | Fig. 5 TEM micrograph of MWCNTs. | |
3.4. Uranium adsorption studies
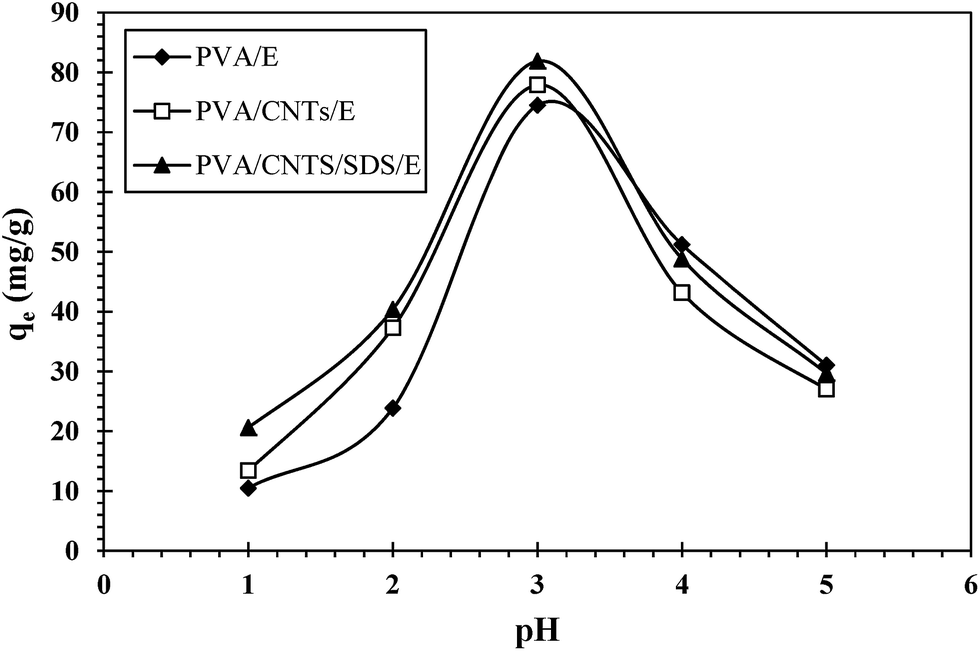
3.4.1. Effect of pH. pH of the aqueous solution is an important parameter that influences metal ion speciation and total surface charge on the chelating resin. The pH effect on UO22+ ions adsorption onto the prepared nanocomposites was investigated for the pH range of 1.0–5.0 at 25 °C. The adsorption capacity as a function of equilibrium pH was illustrated in Fig. 6. It can be seen from Fig. 6 that UO22+ ions adsorption capacity depends on the solution pH value, as the adsorption efficiency increases with pH increasing to a maximum value (pH 3.0) and then decreases with further increase in the solution pH. The maximum adsorption at pH 3 may be due to the formation of U(VI) complexes with carboxyl groups on the surface of MWCNTs and PVA alcoholic groups. At low pH condition (pH < 3.0), the main effective adsorption sites of the hydrogels are easily protonated leading to the reduction of the adsorptive activity.42 However, the availability of free uranium(VI) ions is maximum at pH 3.0 and hence maximum adsorption is obtained.43 With further increase in the solution pH, hydrolysis precipitation starts due to the formation of complexes in aqueous solution. Hydrolysis products such as UO2(OH)+, UO2(OH)22+ and (UO2)3(OH)5+ are formed44 which results in decline of adsorption efficiency of uranium(VI).
 |
| | Fig. 6 Effect of pH on the adsorption of uranium. | |
3.4.2. Effect of contact time. To understand the effect of contact time on UO22+ ions adsorption onto the prepared nanocomposites, adsorption experiments were conducted for contact times ranging from 1 to 200 minute and the obtained results were represented in Fig. 7. The results revealed that UO22+ ions adsorption kinetics had two stages: an initial fast stage where adsorption was fast and contributed significantly to equilibrium uptake, and a slower second phase whose contribution to the total UO22+ ions adsorption was relatively small. The change in the rate of UO22+ ions adsorption might be due to the fact that initially all adsorbent sites were vacant and the solute concentration gradient is high. Afterwards, UO22+ ions adsorption rate decreased significantly due to the decrease in adsorption sites. The quick adsorption of UO22+ ions suggests that chemical adsorption rather than physical adsorption contributes mainly to the UO22+ ions adsorption.45
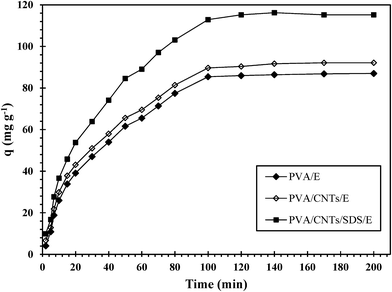 |
| | Fig. 7 Effect of contact time on the adsorption of uranium. | |
3.4.3. Effect of initial concentration. The effect of UO22+ ions initial concentration on the adsorption efficiency was studied by contacting a fixed mass of the prepared nanocomposites at a fixed temperature (25 °C) and pH (3.0) using a range of initial UO22+ ions concentrations (100, 200, 300, 500, 700, and 1000 mg L−1). The adsorption data at different initial UO22+ ions concentrations was shown in Fig. 8. It can be seen that, the adsorption capacities increase with the increment of UO22+ ions initial concentration. This may be due the fact that, the initial concentration provides the driving force to overcome the resistance to the mass transfer of UO22+ ions between adsorbent and adsorbate.46 Meanwhile, the initial concentration was high and then the driving force was high too, and therefore the adsorption capacity would be high.
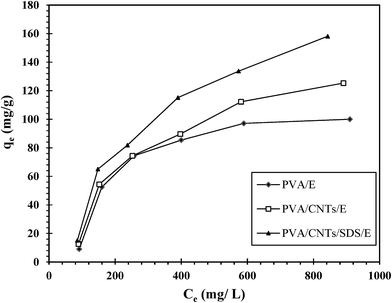 |
| | Fig. 8 Effect of initial concentration on the adsorption of uranium. | |
3.4.4. Evaluation of adsorption isotherm models. In order to understand the adsorption behavior the prepared nanocomposites for UO22+ ions, the equilibrium data were evaluated according to the Langmuir and Freundlich isotherms models under the experimental conditions.Langmuir isotherm model assumes that the adsorbent surface is homogeneous and the adsorption sites are energetically identical indicating that the adsorbed molecules don't react with each other. The linear form of Langmuir equation can be depicted as:47
| |
 | (3) |
where
qe (mg g
−1) represents the amount of solute adsorbed per unit weight of adsorbent at equilibrium;
Ce (mg L
−1) is the equilibrium solute concentration in solution,
Qmax (mg g
−1) is the maximum monolayer adsorption and
b (L mg
−1) the Langmuir constant related to the affinity of binding sites.
The linear form of Freundlich isotherm can be expressed as:48
| |
 | (4) |
where
kF and
n are the Freundlich constants, which represent adsorption capacity and intensity, respectively.
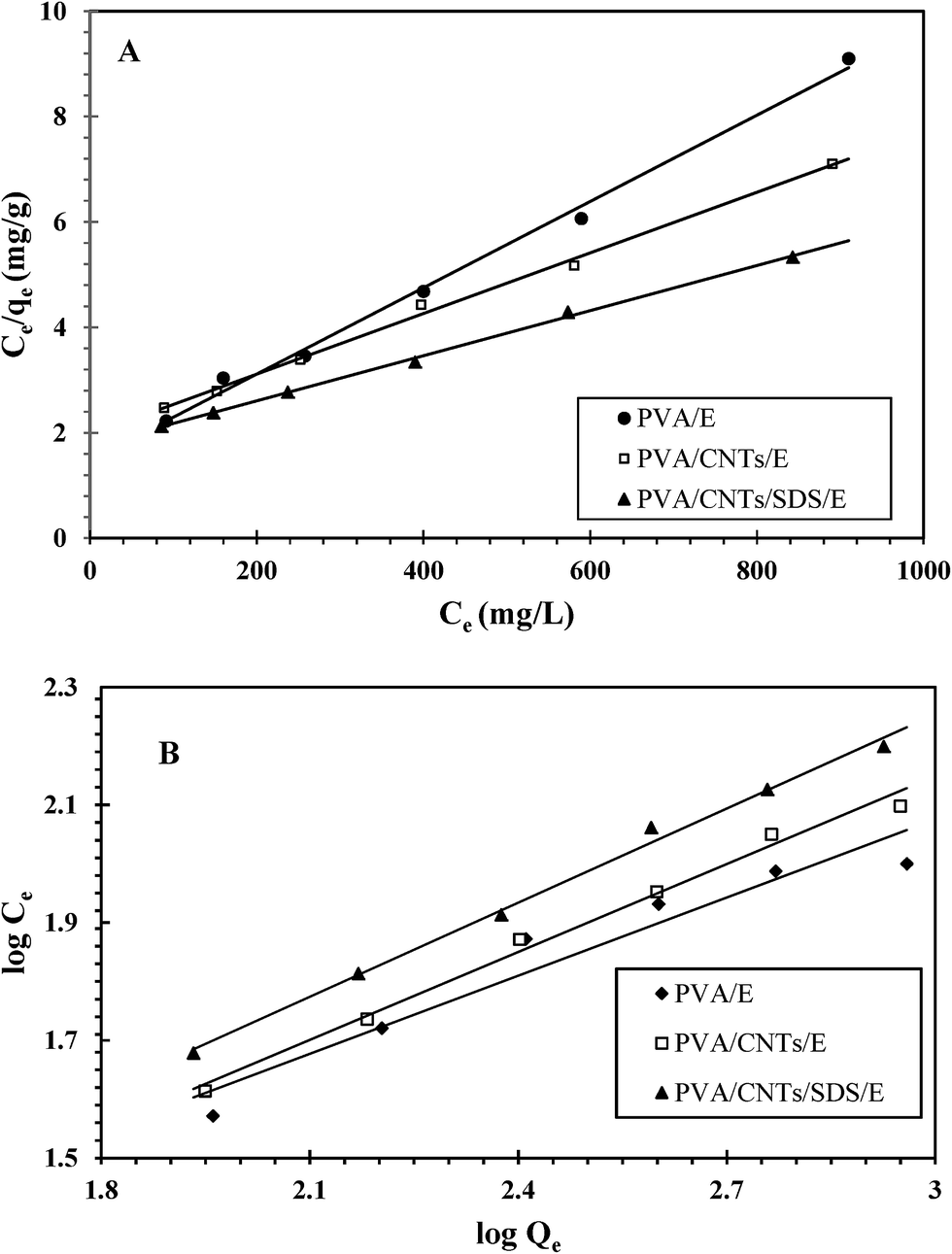
Langmuir and Freundlich sorption isotherms of UO22+ ions on the surface of the prepared hydrogels were elaborated in Fig. 9A and B respectively, and the relative coefficients obtained from fitting isotherm patterns were listed in Table 1. As indicated from the correlation coefficients in Table 1, Langmuir model fits better than Freundlich model for UO22+ ions adsorption on the prepared nanocomposites (correlation coefficient is 0.99). This result suggests that the prepared nanocomposite surface has similar adsorption performance and thus the adsorbed UO22+ ions do not compete with each other and are adsorbed by forming nearly complete monolayer coverage of the nanocomposite particles. This phenomenon suggests that chemo-sorption is the primary adsorption mechanism in adsorption reaction.49 The obtained values of the Langmuir equation parameters specify a high enough adsorption activity of the synthesized sorbent towards uranyl ions.50
 |
| | Fig. 9 Langmuir (A) and Freundlich (B) isotherm plots for the adsorption of uranium. | |
Table 1 Langmuir and Freundlich parameters for uranium(VI) ions adsorption
| Composite |
Langmuir model |
Freundlich model |
| b (L mg−1) |
Q (mg g−1) |
R2 |
kF |
1/n |
R2 |
| PVA/E |
0.0055 |
121.9512 |
0.9928 |
2.7714 |
1.3383 |
0.9318 |
| PVA/CNTs E |
0.0029 |
172.4137 |
0.9933 |
3.1470 |
1.5264 |
0.9890 |
| PVA/CNTs/SDS/E |
0.0024 |
232.5581 |
0.9935 |
3.4127 |
1.5278 |
0.9950 |
Based on the Langmuir equation, Qmax values of UO22+ onto PVA/E, PVA/CNTs/E and PVA/CNTs/SDS/E were 121.95, 172.41 and 232.55 mg g−1 respectively, indicating that the adsorption capacity of the prepared nanocomposite has the following order PVA/E > PVA/CNTs/E > PVA/CNTs/SDS/E. This can be explained as the following: adding MWCNTs to PVA increases the functional groups in the composites consequently the adsorption capacity will increase. It was reported that oxidation of carbon surface can offer not only more hydrophilic surface structure, but also a large number of oxygen-containing functional groups like –COOH, –OH, or –C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O on the surfaces of MWCNTs, which increase the adsorption capability of carbon material.51 The presence of the ionic surfactant, SDS, decreases MWCNTs aggregative tendency in water and enhance their dispersion via noncovalent approach, and therefore enhance the adsorption.52
O on the surfaces of MWCNTs, which increase the adsorption capability of carbon material.51 The presence of the ionic surfactant, SDS, decreases MWCNTs aggregative tendency in water and enhance their dispersion via noncovalent approach, and therefore enhance the adsorption.52
3.5. Adsorption thermodynamics
Thermodynamic parameters including enthalpy change (ΔH°), Gibbs free energy change (ΔG°) and entropy change (ΔS°) were estimated by using equilibrium constants changing with temperature. For this purpose temperature effect on UO22+ ions adsorption was investigated using a water bath with a fixed amount of the prepared nanocomposites at pH 3, and contact time of 180 min for three temperatures: 303, 313 and 323 K. Thermodynamic parameters (ΔH° and ΔS°) for uranium adsorption on the prepared nanocomposites were calculated from the linear plot of lnKd vs. 1/T (Fig. 10), using the following relations:52| |
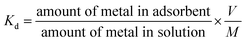 | (5) |
| |
 | (6) |
where Kd is the distribution coefficient (mL g−1), ΔS° is standard entropy, ΔH° is standard enthalpy, T is the absolute temperature (K) and R is the gas constant (J mol−1 K−1).
 |
| | Fig. 10 Plot of lnKd versus 1/T for the adsorption of uranium. | |
The standard free energy values were calculated from the following equation:
Thermodynamic parameters for UO22+ ions adsorption onto the prepared nanocomposites were listed in Table 2. Negative values of ΔH° and the decrease in the value of ΔG° with rise in temperature reveal that the adsorption of UO22+ ions on the prepared nanocomposites is exothermic process in nature and favored at low temperature. Negative free energy values ΔG° indicates the feasibility of the process and its spontaneous nature. Positive values of ΔS° reflect the affinity of the prepared nanocomposites toward UO22+ ions in aqueous solutions and suggest the increased randomness at the solid-solution interface during adsorption.53
Table 2 The thermodynamic parameters for uranium(VI) ions adsorption
| Composite |
ΔH° (kJ mol−1) |
ΔS° (J mol−1 K−1) |
ΔG° (kJ mol−1) |
| PVA/E |
−13.52 |
6.50 |
−11.58 |
−11.43 |
−11.46 |
| PVA/CNTs/E |
−23.79 |
37.09 |
−12.49 |
−12.31 |
−11.74 |
| PVA/CNTs/SDS/E |
−17.26 |
13.80 |
−13.03 |
−13.05 |
−12.75 |
3.6. Desorption analysis
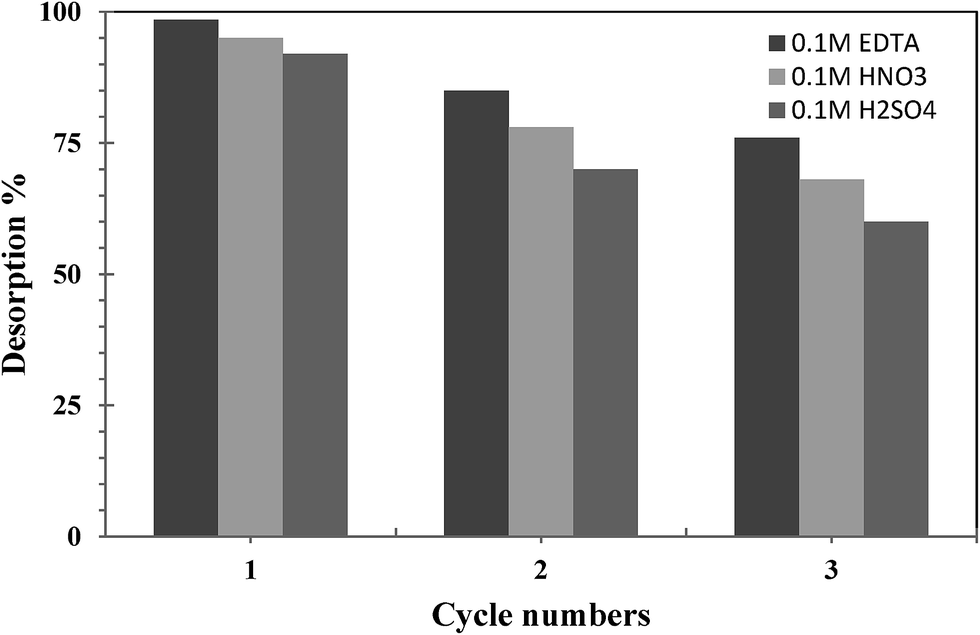
The stability and potential regeneration of the prepared nanocomposites were investigated through desorption. Adsorption/desorption of UO22+ ions onto nanocomposites was studied in a batch mode using H2SO4, HNO3 and EDTA as eluents and the results were represented in Fig. 11. The results indicate that UO22+ ions can be desorbed efficiently by 0.1 mol L−1 EDTA solution. As can be seen from Fig. 11, the adsorption capacity of PVA/MWCNTs nanocomposites towards UO22+ ions decreases slightly from 98.5 to 92.0% after third cycle.
 |
| | Fig. 11 Desorption efficiency of UO22+ ions from the prepared nanocomposite. | |
4. Conclusions
PVA/MWCNTs nanocomposites have been prepared as simple, efficient and feasible UO22+ ions adsorbents. The prepared nanocomposites were characterized by XRD, SEM, TEM and FTIR to determine their chemical constituents, micro-structures and functional groups. UO22+ ions uptake experiments have been carried out by the batch method and the conditions have been optimized. The obtained results show the high adsorption capacity towards UO22+ ions as 121.95, 172.41 and 232.55 mg g−1 for PVA/E, PVA/CNTs/E and PVA/CNTs/SDS/E, respectively. The thermodynamic parameters ΔH°, ΔS° and ΔG° values of UO22+ ions adsorption onto the prepared nanocomposites show that, the process is exothermic and spontaneous in nature. In conclusion, we succeeded to impart the excellent properties of MWCNTs to PVA matrix and to prepare nanocomposite hydrogels which can be used effectively to remove UO22+ ions from aqueous solutions.
References
- E. Craft, A. Abu-Qare, M. Flaherty, M. Garofolo, H. Rincavage and M. Abou-Donia, J. Toxicol. Environ. Health, Part B, 2004, 7, 297–317 CAS.
- S. C. Sheppard, M. I. Sheppard, M. O. Gallerand and B. Sanipelli, J. Environ. Radioact., 2005, 79, 55–83 CrossRef PubMed.
- J. L. Lapka, A. Paulenova, M. Y. Alyapyshev, V. A. Babain, R. S. Herbst and J. D. Law, Radiochim. Acta, 2009, 97, 291–296 CrossRef CAS.
- D. E. Crean, F. R. Livens, M. Sajih, M. C. Stennett, D. Grolimund, C. N. Borca and N. C. Hyatt, J. Hazard. Mater., 2013, 263, 382–390 CrossRef CAS PubMed.
- L. M. Camacho, S. Deng and R. R. Parra, J. Hazard. Mater., 2010, 175, 393–398 CrossRef CAS PubMed.
- S. B. Xie, C. Zhang, X. H. Zhou, J. Yang, X. J. Zhang and J. S. Wang, J. Environ. Radioact., 2009, 100, 162–166 CrossRef CAS PubMed.
- A. M. Atta, Z. H. Abd El Wahab, Z. A. El Shafey, W. I. Zidan and Z. F. Akl, J. Dispersion Sci. Technol., 2010, 31, 1601–1610 CrossRef CAS PubMed.
- S. Zhang, M. Zeng, J. Li, J. Li, J. Xu and X. Wang, J. Mater. Chem. A, 2014, 2, 4391–4397 CAS.
- A. Gajowiak, M. Majdan and K. Drozdzal, Przem. Chem., 2009, 88, 190–196 CAS.
- I. Zhuravlev, O. Zakutevsky, T. Psareva, V. Kanibolotsky, V. Strelko, M. Taffet and G. Gallios, J. Radioanal. Nucl. Chem., 2002, 254, 85–89 CrossRef CAS.
- K. Oshita, M. Oshima, Y. H. Gao, K. H. Lee and S. Motomizu, Anal. Chim. Acta, 2003, 480, 239–249 CrossRef CAS.
- Y. Zhao, J. Li, S. Zhang, H. Chen and D. Shao, RSC Adv., 2013, 3, 18952–18959 RSC.
- Y. Liu, Q. Li, X. Cao, Y. Wang, X. Jiang, M. Lee, M. Hua and Z. Zhang, Appl. Surf. Sci., 2013, 285, 258–266 CrossRef CAS PubMed.
- P. Liang, Y. C. Qin, B. Hu, T. Y. Peng and Z. C. Jiang, Anal. Chim. Acta, 2001, 440, 207–213 CrossRef CAS.
- S. Zhang, J. Li, X. Wang, Y. Huang, M. Zeng and J. Xu, J. Mater. Chem. A, 2015, 3, 10119–10126 CAS.
- S. Zhang, J. Li, X. Wang, Y. Huang, M. Zeng and J. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 22116–22125 CAS.
- M. Cui, J. Huang, Y. Wang, Y. Wu and X. Luo, Biosens. Bioelectron., 2015, 68, 563–569 CrossRef CAS PubMed.
- S. Prabhu and E. K. Poulose, Int. Nano Lett., 2012, 2, 32–33 CrossRef.
- S. Zhang, J. Li, X. Wang, Y. Huang, M. Zeng and J. Xu, J. Mater. Chem. A, 2013, 1, 11691–11697 CAS.
- X. Wang, Y. Guo, L. Yang, M. Han, J. Zhao and X. Cheng, J. Environ. Anal. Toxicol., 2012, 2, 154–161 Search PubMed.
- J. Wang, P. Liu, Z. Li, W. Qi, Y. Lu and W. Wu, Materials, 2013, 6, 4168–4185 CrossRef CAS PubMed.
- A. Gopalan, M. F. Philips, J. H. Jeong and K. P. Lee, J. Nanosci. Nanotechnol., 2014, 14, 2451–2458 CrossRef CAS PubMed.
- J. N. Dawoud, Appl. Surf. Sci., 2012, 259, 433–440 CrossRef PubMed.
- V. V. Didenko, V. C. Moore, D. S. Baskin and R. E. Smalley, Nano Lett., 2005, 5, 1563–1567 CrossRef CAS PubMed.
- R. B. Mathur, S. Seth, C. Lal, R. Rao, B. P. Singh and T. L. Dhami, Carbon, 2007, 45, 132–140 CrossRef CAS PubMed.
- Z. Wang, H. A. Colorad, Z.-H. Guo, H. Kim, C.-L. Park, H. Tomas Hahn, S.-G. Lee, K.-H. Lee and Y. Qin, Mater. Res., 2012, 15, 510–516 CrossRef CAS.
- B. S. Stankovich, D. A. Dikin, R. D. Piner, K. A. Kohlhaas, A. Kleinhammes, Y. Jia, Y. Wu, S. T. Nguyen and S. Rodney, Carbon, 2007, 45, 1558–1565 CrossRef PubMed.
- Z. Abdeen, J. Dispersion Sci. Technol., 2011, 32, 1337–1344 CrossRef CAS PubMed.
- J. Jose, S. K. De, M. Al-Ali, J. B. Dakua, P. A. Sreekumar, R. Sougrat and M. A. Al-Harthi, Starch, 2015, 67, 147–153 CrossRef CAS PubMed.
- P. Zhang, T. Zhou, L. He, S. J. Sun, J. Wang, C. Qin and L. Dai, RSC Adv., 2015, 5, 55492–55498 RSC.
- P. Zhang, D. Qiu, H. Chen, J. Sun, J. Wang, C. Qin and L. Dai, J. Mater. Chem. A, 2015, 3, 1442–1449 CAS.
- C. W. Lou, Z. I. Lin, C. L. Huang, W. C. Chen, C. K. Chen and J. H. Lin, Appl. Mech. Mater., 2015, 749, 182–185 CrossRef.
- F.-P. Du, E.-Z. Ye, W. Yang, T.-H. Shen, C.-Y. Tang, X.-L. Xie, X.-P. Zhou and W.-C. Law, Composites, Part B, 2015, 68, 170–175 CrossRef CAS PubMed.
- F. T. Chi, S. Hu, J. Xiong and X. Wang, Sci. China: Chem., 2013, 56, 1495–1503 CrossRef CAS.
- Y. Liu, X. Caoc, R. Huac, Y. Wang, Y. Liu, C. Pang and Y. Wang, Hydrometallurgy, 2010, 104, 150–155 CrossRef CAS PubMed.
- S. T. Yang, W. Guo, Y. Lin, X. Y. Deng, H. F. Wang, H. F. Sun, Y. F. Liu, X. Wang, W. Wang, M. Chen, Y. P. Huang and Y. P. Sun, J. Phys. Chem. C, 2007, 111, 17761–17764 CAS.
- A. M. AL-Sabagh and Z. Abdeen, J. Polym. Environ., 2010, 18, 576–583 CrossRef CAS.
- L. Liu, A. H. Barber, S. Nuriel and H. D. Wanger, Adv. Funct. Mater., 2005, 15, 975–980 CrossRef CAS PubMed.
- J. Liang, Y. Huang, L. Zhang, Y. Wang, Y. Ma, T. Guo and Y. Chen, Adv. Funct. Mater., 2009, 19, 2297–2302 CrossRef CAS PubMed.
- Y. Kim, N. Minami and S. Kazaoui, Appl. Phys. Lett., 2005, 86, 073103 CrossRef PubMed.
- M. Kokabi, M. Sirousazar and Z. M. Hassan, Eur. Polym. J., 2007, 43, 773–781 CrossRef CAS PubMed.
- Y. H. F. Al-qudah, G. A. Mahmoud and M. A. Abdel Khalek, J. Radiat. Res. Appl. Sci., 2014, 7, 135–145 CrossRef CAS PubMed.
- G. Wanga, J. Liu, X. Wang, Z. Xie and N. Deng, J. Hazard. Mater., 2009, 168, 1053–1058 CrossRef PubMed.
- P. Ilaiyaraja, A. Kumar Singh Deba, K. Sivasubramaniana, D. Ponrajub and B. Venkatramana, J. Hazard. Mater., 2013, 250–251, 155–166 CrossRef CAS PubMed.
- S. Yang, J. Li, D. Shao, J. Hu and X. Wang, J. Hazard. Mater., 2009, 166, 109–116 CrossRef CAS PubMed.
- G. Bayramoglu, B. Altintas and M. Y. Arica, Chem. Eng. J., 2009, 152, 339–346 CrossRef CAS PubMed.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1368 CrossRef CAS.
- H. Freundlich, Z. Phys. Chem., 1906, 57, 384–470 Search PubMed.
- G. D. Sheng, L. Ye, Y. M. Li, H. P. Dong, H. Li, X. Gao and Y. Y. Huang, Chem. Eng. J., 2014, 248, 71–78 CrossRef CAS PubMed.
- E. O. Akperov, A. M. Maharramov and O. G. Akperov, Hydrometallurgy, 2009, 100, 76–81 CrossRef CAS PubMed.
- C. Chen, X. Li, D. Zhao, X. Tan and X. Wang, Colloids Surf., A, 2007, 302, 449–454 CrossRef CAS PubMed.
- L. Vaisman, H. D. Wagner and G. Marom, Adv. Colloid Interface Sci., 2006, 128–130, 37–46 CrossRef CAS PubMed.
- S. Chen, J. Hong, H. Yang and J. Yang, J. Environ. Radioact., 2013, 126, 253–258 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.