DOI:
10.1039/C5RA10156E
(Paper)
RSC Adv., 2015,
5, 65991-65997
Ammonia borane in an external electric field: structure, charge transfer, and chemical bonding†
Received
29th May 2015
, Accepted 24th July 2015
First published on 24th July 2015
Abstract
It is well known that ammonia borane (BH3NH3) is one of the simplest donor–acceptor complexes. The donor–acceptor bond (B–N bond) is formed by sharing lone pair electrons between BH3 and NH3 groups. In the present work, different strengths of external electric fields (Eext) are applied along the Z-axis direction to investigate the electric field induced effect on the BH3NH3 structure and properties. Interestingly, we have found that the lone pair electrons can be gradually moved in the “channel” between the BH3 and NH3 groups by modulating Eext. The donor–acceptor bond (B–N bond) is gradually elongated until it breaks when the Eext ranges from 0.0000 to 0.0321 au. The B–N bond is the shortest at Eext = −0.0519 au. Interestingly, the negative charge on BH3 groups sharply decreases from 0.161 to 0.005 (for NH3 groups decreases from 0.161 to 0.005) and the electron cloud of HOMO−2 exhibits an obvious transformation at “broking bonding” Eext ranging from 0.0320 to 0.0321 au, indicating the electron movement induced by the electric-field is the main reason to change the structure and stability of BH3NH3. Further, atoms in molecules (AIM) analysis shows that the B⋯N interactions are similar to that of hydrogen bonds at Eext = −0.0767 au and the iconicity of B–N bond in BH3NH3 is confirmed by its low electron density for B–N ρ(B–N) (0.103–0.073) in the region of Eext (0–0.025 au).
1. Introduction
Since the synthesis and isolation of famous ammonia borane (BH3NH3) was realized by Shore and Parry,1 its structure,2 phase transition,2i,3 hydrogen storage3d,4 and dehydrogenation5 have been investigated by numerous experimental or theoretical studies. It is well known that BH3NH3 is one of the simplest electron donor–acceptor complexes2b which is formed by the dative bond between the electron rich NH3 as the Lewis acid and electron deficient BH3 as the Lewis base.2g The variations in the geometrical and electronic characteristics of the isolated BH3NH3 molecule along the donor–acceptor distance (dB–N) have been analyzed at the MP2(fc)/6-311+G(2df,p) level.2c The boron–nitrogen dative bond (B–N bond) is formed by electron transfer from the lone nitrogen pairs to the vacant orbital on boron2e,f and B–N bond distance is 1.658 Å for the isolated BH3NH3 molecule which is inferred from a microwave spectrum.2b,h However, for the crystal structure, the B–N bond distance is 1.597 Å which is reported by single crystal X-ray spectroscopy at 298 K.2h,i The shortening of B–N distance can be attributed to the dipole–dipole interaction between “middle” BH3NH3 molecule and surrounding other BH3NH3 units.6 As a white crystalline solid at room temperature, BH3NH3 can be transformed into ordered orthorhombic structure at low-temperature and a transition to tetragonal structure accurse around 255 K.3b The investigation of bonding in low-temperature orthorhombic phase of BH3NH3 finds the existence of a strong covalent bond between B–H atoms.2d Significantly, hydrogen is a high chemical stable and hydrogen content (19.6 wt%)3d,7 compound which is considered as an important hydrogen storage material. In addition, hydrogen release from BH3NH3 and its dimers can be controlled by modulated temperature.2h,4b,8 Therefore, it is has been discovered as potential materials in fuel cell application.4a,5a,7b,9 The synthesis of the compound bis-BN cyclohexane which is an unusually kinetically stable chemical hydrogen storage material with H2 storage capacity of 4.7 wt% have been developed.10
On the other hand, the effect of an electric field is widely applied in the field of controlling and modulating the electric and magnetic properties of organic conductors,11 hydrogen bonding complexes,12 nanotubes,13 and graphene,14 and so on. Li group found that the degree of proton transfer for HN3–HCI and H2O–HCl can be controlled by modulating the strength of Eext.12 The application of a static external electric field have resulted in the enhancement of third-order nonlinear optical (NLO) response in symmetric diradicals.15 In addition, our group has investigated the second-order NLO response of HArF system optimized after different external electric field.16
From the above discussion, we can find that the formation of B–N bond may be ascribed to donation of the ammonia lone-pair electrons (see Scheme 1), and external electric field can be applied to control structures and properties of a certain matter. In present work, the Eext is applied along Z-axis direction of BH3NH3 to control its electron movement, molecular structure and chemical bonding characteristic. It is our hope that this work may provide a new method to modulate the stability of BH3NH3 by controlling structure of donor–acceptor complex BH3NH3.
2. Computational details
The MP2 method have been applied to investigate the structure and properties of BH3NH3, and the equilibrium geometrical parameters at the MP2/6-311+G(2df,p) level are close to experimental geometry.2c,g Therefore, the optimized geometric structures of BH3NH3 with all real frequency in a series of external electric fields −0.0767 to 0.0321 au (1 au = 5.142 × 109 V cm−1) were obtained at the level of MP2/6-311+G(2df,p). The external electric field was applied along Z-axis direction which is parallel to B–N bond. The natural bond orbital (NBO) analyses were performed at the QCISD/aug-cc-pVTZ level and the NBO population of BH3NH3 in/after different external electric fields has been listed in Table S2.† The topological properties of the electron density distribution were studied by atoms in molecules (AIM)17 theory at MP2/6-311+G(2df,p) level. In addition, the optimized structural parameters of (BH3NH3)2 dimers in a series of external electric fields (−0.0200 to 0.0200 au) were also obtained at the level of MP2/6-311+G(2df,p). The external electric field direction is parallel to B–N bond. All of the calculations were performed with Gaussian 09 software package.18
3. Results and discussion
3.1 The structures of BH3NH3 in external electric field
The selected structural parameters of BH3NH3 in external electric fields have been listed in Table 1, and others are listed in Table S1 of the ESI.† When Eext along Z-axis negative direction is applied to BH3NH3 and gradually increase from 0.0000 to 0.0519 au, the B–N bond is slowly shrinked from 1.649 to 1.606 Å (see Fig. 1 and Table 1). This decrease of B–N distance suggests that the interaction between B atom and N atom slowly gets stronger and the stability of BH3NH3 can be increased. The increased stability further indicates we can modulate the degree of hydrogen storage by controlling strength of Eext along Z-axis negative direction. However, when Eext further increases from 0.0519 to 0.0767 au, the B–N bond is gradually elongated by 0.047 Å. This change indicates that the interaction between B and N atom slowly becomes weak, and the BH3 groups and NH3 groups tend to be separated from each other. On the other hand, the external electric field is applied along Z-axis positive direction and Eext increases from 0.0000 to 0.0321 au. From Fig. 1 and Table 1, under the region of Eext (0.0000–0.0320 au), it can be obviously found that B–N distance is gradually elongated from 1.649 to 1.923 Å and the stability of BH3NH3 is gradually weaker and weaker. This variation of stability suggests that our work may provide a new method to modulate the degree of hydrogen release. After this, when Eext further increases from 0.0320 to 0.0321 au, an interesting phenomenon can be found: B–N distance is elongated suddenly by 1.305 Å which is 5 times larger than the increment 0.253 Å (Eext is from 0.0000 to 0.0320 au). This sudden crucial sharp increase of B–N distance apparently indicates that the B–N bond is suddenly broken at Eext = 0.0321 au. The adjustable stability of BH3NH3 shows that our work may provide a potential method to control the degree of hydrogen release or storage.
Table 1 The optimized structural parameters and dipole moment (μ) of BH3NH3 determined at the MP2/6-311+G(2df,p) level in some important external electric fieldsa
| Eext × 10−4 au |
r(B–N) |
r(B–H) |
r(N–H) |
H–B–H |
H–N–H |
H1–B–H2–H3 |
H′1–N–H′2–H′3 |
μ1 |
μ2 |
| The bond distance are in angstroms, angles; in degrees; dipole moment, in Debye. |
| −767 |
1.653 |
1.263 |
1.045 |
106.7 |
101.0 |
113.9 |
103.7 |
13.33 |
6.30 |
| −519 |
1.606 |
1.239 |
1.031 |
108.1 |
102.8 |
116.8 |
106.5 |
10.16 |
6.14 |
| 0 |
1.649 |
1.208 |
1.017 |
113.6 |
107.9 |
131.9 |
116.3 |
5.44 |
5.44 |
| 320 |
1.902 |
1.197 |
1.009 |
118.3 |
114.9 |
154.7 |
136.8 |
1.80 |
3.97 |
| 321 |
3.207 |
1.192 |
1.021 |
119.7 |
102.3 |
168.3 |
105.7 |
4.20 |
2.01 |
 |
| | Fig. 1 Variations of B–N distance with the increase of Eext. | |
In order to further clearly show structural variations of BH3NH3, the B–H bond distance and dihedral angle H1–B–H2–H3 of BH3 groups (N–H bond distance and dihedral angle H′1–N–H′2–H′3 of NH3 groups) in different external electric fields have been analyzed, respectively. The trends of the dihedral angle H1–B–H2–H3 and H′1–N–H′2–H′3 are shown in Fig. 2. For NH3 groups (see Fig. 2 and Table 1), in the range of Eext (0.0000–0.0320 au), the dihedral angle H′1–N–H′2–H′3 gradually increases from 116.3 to 136.8° which indicates that structure of NH3 groups gradually tend to be coplanar. However, when Eext further increases to 0.0321 au, dihedral angle H′1–N–H′2–H′3 suddenly decreases to 105.7° which indicates that NH3 groups still has a trigonal pyramidal shape at Eext = 0.0321 au. For BH3 groups (see Fig. 2 and Table 1), when the Eext gradually increase from 0.0000 to 0.0321 au, the B–H bond is shrinked from 1.208 to 1.192 Å and the values of dihedral angle H1–B–H2–H3 increase from 131.9 to 168.3°. As we all known, the structure of borane (BH3) is trigonal planar (D3h molecular symmetry) with experimentally determined by the B–H bond length of 119 pm.19 Hence, it can be obviously found that the shape of BH3 groups gradually tends to be coplanar with the increasing of Eext along Z-axis positive direction.
 |
| | Fig. 2 Variation of dihedral angle H1–B–H2–H3 and H′1–N–H′2–H′3 with the increase of Eext. | |
The optimized structural parameters of (BH3NH3)2 dimers in different external electric fields have been shown in Table S4 of the ESI.† From Table S4,† we can find that the B–N bond of BH3NH3 monomer is longer than (BH3NH3)2 dimmers, indicating that the electron correlation have effected B–N bond length. In the region of Eext (0.0000–0.0100 au), the B–N bond is gradually elongated and the B–N bond length are all shorter than 1.649 Å. This change suggests that the effects of electron correlations are stronger than the Eext. However, when the Eext increases to 0.0150 au, the B–N bond length is longer than 1.649 Å, indicating that the effect of Eext on B–N bond is more obvious than electron correlations. To explain the variations of the structure of BH3NH3 in different external electric fields, we further analyze the charge transfer between BH3 and NH3 groups.
3.2 The charge transfer analysis
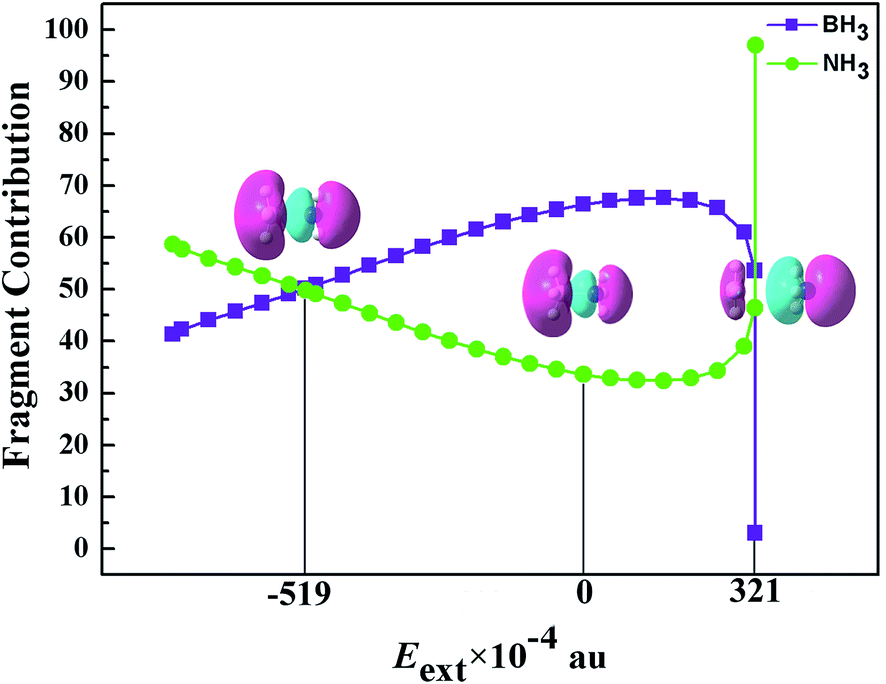
A The valence molecular orbital of BH3NH3. The formation of B–N bond has been explained by the interaction of occupied molecular orbital of NH3 and unoccupied molecular orbital of BH3.20 The boron atom having three valence is subject to sp-orbital hybridization with an empty orbital to interact with lone nitrogen pairs through electrostatic interaction. The valence molecular orbital of BH3NH3 in some important Eext has been shown in Fig. 3. From Fig. 3, we can easily find that the linear combinations of the lowest unoccupied molecular orbital (LUMO) of BH3 groups and the highest occupied molecular orbital (HOMO) of NH3 groups constitute the bonding orbital (HOMO−2) and the antibonding orbital (LUMO+3) of BH3NH3. When the external electric field is applied along the Z-axis positive direction and Eext gradually increases from 0.0000 to 0.0320 au, the electron cloud of HOMO−2 is not obviously shifted. However, when Eext further increases from 0.0320 to 0.0321 au, the electron cloud of HOMO−2 exhibits a dramatic transformation. With the increase of Eext, the electric cloud of HOMO−2 is more and more concentrated on NH3 groups, which is in agreement with the trend of B–N distance and dihedral angle H1–B–H2–H3 (or H′1–N–H′2–H′3). It is shown that the lone nitrogen pairs of NH3 groups can be moved along electric field direction by modulating Eext. Then, the electrostatic interaction of B–N bond are modulated, which result in the elongation of B–N distance and the change of dihedral angle dihedral angle H1–B–H2–H3 (or H′1–N–H′2–H′3).
 |
| | Fig. 3 The molecular orbitals of BH3NH3 in external electric field. | |
B Dipole moment. The dipole moment of BH3NH3 at the MP2/6-311+G(2df,p) level are listed in Table 1 and Table S1.† The variations of the dipole moment (μ1) of BH3NH3 in external electric field and the dipole moment (μ2) of BH3NH3 after external electric field have been shown in Fig. 4. From Fig. 4, it is obvious that the values of μ1 and μ2 all gradually decrease when the Eext is from −0.0767 to 0.0320 au. Most importantly, the values of μ1 are different from μ2 in the same Eext, and the difference of μ1 and μ2 values in the same Eext are gradually increase when the Eext increase from 0 to −0.0767 au. It suggests that the movement of electrons caused by the external electric field results in the change of dipole moment.
 |
| | Fig. 4 Variations of dipole moment (μ1) of BH3NH3 in external electric fields and dipole moment (μ2) of BH3NH3 after external electric fields. | |
C Natural bond orbital (NBO) analyses. The natural bond orbital (NBO) analyses of BH3NH3 at the MP2/6-311+G(2df,p) level are listed in Table 2 and Table S2.† The variations of NBO analyses of BH3NH3 in/after external electric field have been shown in Fig. 5. From Fig. 5 and Table 2, we can obviously find that the charge locates at BH3 and NH3 groups of BH3NH3 after external electric field have no obvious change when the Eext is from −0.0767 to 0.0320 au. However, for BH3NH3 in external electric field along Z-axis positive direction, the positive charge on NH3 groups gradually decreases from 0.316 (Eext = 0 au) to 0.161 (Eext = 0.0320 au) and the negative charge on BH3 groups decreases from 0.316 (Eext = 0 au) to 0.161 (Eext = 0.0320 au). After this, when Eext increases from 0.0320 au to 0.0321 au (see Fig. 5), the negative charge on BH3 groups suddenly decreases from 0.161 to 0.005 (for NH3 groups is from 0.161 to 0.005). Significantly, the variations of NBO charge population are exactly accordance with the elongation of B–N bond length and the change of dihedral angle H1–B–H2–H3 (or H1′–N–H′2–H′3). It is shown that the electron movement from BH3 groups to NH3 groups results in the change of electrostatic interaction. And then, the B–N bond is gradually elongated and the shapes of BH3 groups and NH3 groups show tends to be coplanarity, respectively. When the BH3 groups and NH3 groups separated from each other (Eext = 0.0321 au), the gradually decreased interaction lead to the shape of BH3 groups is nearly coplanar and NH3 groups reform its trigonal pyramidal shape. On the other hand, by adding external electric field along Z-axis negative direction, charge on NH3 groups gradually increases from 0.316 (Eext = 0 au) to 0.706 (Eext = 0.0767 au) and the negative charge on BH3 groups increase from 0.316 (Eext = 0 au) to 0.706 (Eext = 0.0320 au). Obviously, lone nitrogen pairs of NH3 groups are gradually moved to electron-deficient BH3 groups to form stable octet configuration. Due to the existence of the repelling interaction, this electron movement results in slow enhance of interaction between BH3 groups and NH3 groups. Therefore, the B–N bond is slowly shrinked when the Eext along Z-axis negative direction increases in appropriate range. In addition, the partial negative charges on N atom show very special trend which increase from 0.911 (Eext = −0.0767 au) to 1.18 (Eext = 0.0321 au) and the charge on B atom increase from −0.186 to 0.432. How do these interesting electron movement affect the chemical bonding in BH3NH3?
Table 2 NBO charge q1 of BH3NH3 in external electric fields and NBO charge q2 of BH3NH3 after external electric fields
| Eext × 10−4 au |
q1(NH3) |
q1(BH3) |
q1(N) |
q1(B) |
q2(NH3) |
q2(BH3) |
| −767 |
0.706 |
−0.706 |
−0.911 |
−0.186 |
0.318 |
−0.318 |
| −519 |
0.449 |
−0.449 |
−0.997 |
0.044 |
0.329 |
−0.329 |
| 0 |
0.316 |
−0.316 |
−0.935 |
0.021 |
0.313 |
−0.313 |
| 320 |
0.161 |
−0.161 |
−1.033 |
0.162 |
0.237 |
−0.237 |
| 321 |
0.005 |
−0.005 |
−1.180 |
0.432 |
0.018 |
−0.017 |
 |
| | Fig. 5 The NBO charge of BH3 groups and NH3 groups in BH3NH3. | |
3.3 The chemical bonding analysis
To analyze the chemical bonding of BH3NH3 under a series of Eext, atoms in molecules (AIM) analysis is performed by using the calculation results at MP2/6-311+G(2df,p) level. The results of the electron density ρ and the Laplacian ∇2ρ(r) at the bond critical points (BCP) have been shown in Tables 3 and S2.† The Laplacian of the electron density and electron density ρ of the bond critical points (BCP) have been used to analyze the types of chemical bonds.2a,21 For electrostatic interaction (ionic bond, hydrogen bonds, and van der Waals interactions) the Laplacian of the electron density ∇2ρ(r) is positive, while the ∇2ρ(r) is negative for the covalent bond. The ∇2ρ(r) value of hydrogen bond lies in the proposed range of 0.014–0.139 au. From Table 3, in the range of −0.0767 to 0.0250 au, we obviously find that the ∇2ρ(r) for B–N is from 0.124 to 0.436 indicating that the B–N bond is electrostatic interaction. When the Eext is from 0 to 0.0250 au, the iconicity of B–N bond in BH3NH3 is confirmed by its low ρ(B–N) (0.103–0.073).2a In addition, it is obvious that the value (0.134) of ∇2ρ(r) for B–N lies in the range of 0.014–0.139 au when Eext is 0.0767 au. This interesting phenomenon indicates that the B⋯N interaction are similar to that of hydrogen bonds at Eext = −0.0767 au. It is well known that covalent bonding is observed between B–H atom (N–H atom) in BH3NH3.2d From Table 3, without external electric field, the negative values of ∇2ρ(r) for B–H and ∇2ρ(r) for N–H just demonstrate that covalent bond exists between B–H atom (N–H atom). Furthermore, in the region of −0.0767 to 0.0321 au, the negative values of ∇2ρ(r) for B–H and ∇2ρ(r) for N–H confirm that the B–H bond and N–H bond are always covalent bond when different external electric fields are applied to BH3NH3. In addition, the detailed fragment contributions to the bonding orbital (HOMO−2) of the BH3NH3 have been analyzed and shown in Fig. 6.
Table 3 The Laplacian of the electron density (∇2ρ(r)) and electron density (ρ) of the bond critical points (BCPs)
| Eext × 10−4 au |
∇2ρ(B–N) |
∇2ρ(B–H) |
∇2ρ(N–H) |
ρ(B–N) |
ρ(B–H) |
ρ(N–H) |
| −767 |
0.124 |
−0.053 |
−1.809 |
0.134 |
0.140 |
0.310 |
| −519 |
0.314 |
−0.063 |
−1.828 |
0.136 |
0.151 |
0.314 |
| 0 |
0.494 |
−0.128 |
−1.775 |
0.103 |
0.169 |
0.342 |
| 50 |
0.494 |
−0.137 |
−0.137 |
0.099 |
0.170 |
0.343 |
| 100 |
0.495 |
−0.146 |
−0.146 |
0.094 |
0.172 |
0.343 |
| 150 |
0.487 |
−0.155 |
−0.155 |
0.088 |
0.173 |
0.344 |
| 200 |
0.469 |
−0.167 |
−0.167 |
0.081 |
0.174 |
0.344 |
| 250 |
0.436 |
−0.180 |
−0.180 |
0.073 |
0.176 |
0.344 |
| 300 |
|
−0.200 |
−0.200 |
|
0.178 |
0.344 |
| 320 |
|
−0.217 |
−1.775 |
|
0.180 |
0.344 |
| 321 |
|
−0.288 |
−1.621 |
|
0.186 |
0.335 |
 |
| | Fig. 6 The detailed fragment contribution to the bonding orbital HOMO−2 of BH3NH3. | |
In order to further reveal the relation between the electric structure and geometric structure for BH3NH3, the localized orbital locator22 (LOL) and electron location function23 (ELF) analyses over BH3NH3 were carried out. Fig. 7 shows the LOL and ELF schemes of BH3NH3 in some important external electric fields. For BH3NH3, both the LOL and ELF maps indicate that the significant coordination electron density localization between N atom and B atom get weaker and weaker when the Eext increases from 0 au to 0.321 au. Most importantly, electron density localization is not observed between N atom and B atom, indicating bond breaking when the external electric field increases to 0.0321 au. These results clearly explain the increase of B–N distance when the external electric field along Z-axis positive is applied to BH3NH3.
 |
| | Fig. 7 The ELF and LOL models of BH3NH3 in different external electric fields. | |
 |
| | Scheme 1 Chemical structure of BH3NH3. | |
4. Conclusions
In present work, we added a series of different strength of external electric fields along Z-axis direction of BH3NH3 to investigate the electric field induced effect on electron transfer, structure and properties of BH3NH3. Interestingly, when the Eext along Z-axis positive direction increase from 0 to 0.0321 au, the B–N bond is gradually elongated until broken at Eext = 0.0321 au. The change of the B–N bond distance indicates the stability can be modulated by adding Eext. With the increase of Eext along Z-axis positive direction, the electric cloud of HOMO−2 is more and more concentrated on NH3 groups and the positive charge on NH3 groups decreases from 0.316 (Eext = 0.0320 au) to 0.005 (Eext = 0.0321 au) and the negative charge BH3 groups decreases from 0.316 (Eext = 0.0320 au) to 0.005 (Eext = 0.0321 au). The variations of NBO charge are in agreement with the trend of B–N distance, which indicates the electrostatic interaction of B–N bond are modulated by controlling electron movement induced by electric-field. From the atoms in AIM analysis, in the range of Eext (0–0.0250 au), the iconicity of B–N bond in BH3NH3 is confirmed by its low ρ(B–N) (0.103–0.073). In addition, the B⋯N interaction are similar to that of hydrogen bonds at Eext = −0.0767 au due to the value (0.134) for ∇2ρ(r) for B–N lies in the range of 0.014–0.139 au. As such, our work may put forward a new method to control the structure and chemical bonding characteristic for donor–acceptor complex BH3NH3.
Acknowledgements
The authors gratefully acknowledge financial support from National Science Foundation of China (NSFC) (21473026), the Science and Technology Development Planning of Jilin Province (20140101046JC), and H.-L. X. acknowledges support from the Hong Kong Scholars Program XJ2013007. And Project funded by China Postdoctoral Science Foundation (2014M560227).
References
-
(a) S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 8–12 CrossRef CAS;
(b) S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1955, 77, 6084–6085 CrossRef CAS.
-
(a) P. L. A. Popelier, J. Phys. Chem. A, 1998, 102, 1873–1878 CrossRef CAS;
(b) L. R. Thorne, J. Chem. Phys., 1983, 78, 167–171 CrossRef CAS PubMed;
(c) V. Horváth, A. Kovács and I. Hargittai, J. Phys. Chem. A, 2003, 107, 1197–1202 CrossRef;
(d) B. Lingam Ch, K. R. Babu, S. P. Tewari and G. Vaitheeswaran, J. Comput. Chem., 2011, 32, 1734–1742 CrossRef PubMed;
(e) J. S. Binkley and L. R. Thorne, J. Chem. Phys., 1983, 79, 2932–2940 CrossRef CAS PubMed;
(f) L. S. Fisher, K. McNeil, J. Butzen and T. A. Holme, J. Phys. Chem. B, 2000, 104, 3744–3751 CrossRef CAS;
(g) P. E. M. Siegbahn, O. Eisenstein, A. L. Rheingold and T. F. Koetzle, Acc. Chem. Res., 1996, 29, 348–354 CrossRef PubMed;
(h) A. Staubitz, A. P. M. Robertson and I. Manners, Chem. Rev., 2010, 110, 4079–4124 CrossRef CAS PubMed;
(i) M. E. Bowden, G. J. Gainsford and W. T. Robinson, Aust. J. Chem., 2007, 60, 149–153 CrossRef CAS.
-
(a) J. Nylén, L. Eriksson, D. Benson and U. Häussermann, J. Chem. Phys., 2013, 139, 054507 CrossRef PubMed;
(b) C. F. Hoon and E. C. Reynhardt, J. Phys. C: Solid State Phys., 1983, 16, 6129–6136 CrossRef CAS PubMed;
(c) N. J. Hess, M. E. Bowden, V. M. Parvanov, C. Mundy, S. M. Kathmann, G. K. Schenter and T. Autrey, J. Chem. Phys., 2008, 128, 034508 CrossRef PubMed;
(d) J. B. Yang, J. Lamsal, Q. Cai, W. J. James and W. B. Yelon, Appl. Phys. Lett., 2008, 92, 091916 CrossRef PubMed.
-
(a) N. C. Smythe and J. C. Gordon, Eur. J. Inorg. Chem., 2010, 509–521 CrossRef CAS PubMed;
(b) F. H. Stephens, V. Pons and R. Tom Baker, Dalton Trans., 2007, 2613–2626 RSC;
(c) A. Gutowska, L. Li, Y. Shin, C. M. Wang, X. S. Li, J. C. Linehan, R. S. Smith, B. D. Kay, B. Schmid, W. Shaw, M. Gutowski and T. Autrey, Angew. Chem., Int. Ed., 2005, 44, 3578–3582 CrossRef CAS PubMed;
(d) T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116–8118 CrossRef CAS PubMed;
(e) Z. Tang, X. Chen, H. Chen, L. Wu and X. Yu, Angew. Chem., Int. Ed. Engl., 2013, 52, 5832–5835 CrossRef CAS PubMed.
-
(a) Y. Kim, Y. Kim, S. Yeo, K. Kim, K. Jung-Eun Koh, J.-E. Seo, S. J. Shin, D.-K. Choi, C. W. Yoon and S. W. Nam, J. Power Sources, 2013, 229, 170–178 CrossRef CAS PubMed;
(b) M. C. Denney, V. Pons, T. J. Hebden, D. M. Heinekey and K. I. Goldberg, J. Am. Chem. Soc., 2006, 128, 12048–12049 CrossRef CAS PubMed;
(c) T. Umegaki, A. Seki, Q. Xu and Y. Kojima, J. Alloys Compd., 2014, 588, 615–621 CrossRef CAS PubMed.
- M. P. Mitoraj, J. Phys. Chem. A, 2011, 115, 14708–14716 CrossRef CAS PubMed.
-
(a) C. R. Miranda and G. Ceder, J. Chem. Phys., 2007, 126, 184703 CrossRef PubMed;
(b) F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., Int. Ed., 2007, 46, 746–749 CrossRef CAS PubMed;
(c) W. T. Klooster, T. F. Koetzle, P. E. M. Siegbahn, T. B. Richardson and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 6337–6343 CrossRef CAS.
- S. Swinnen, V. S. Nguyen and M. T. Nguyen, Chem. Phys. Lett., 2011, 517, 22–28 CrossRef CAS PubMed.
- B. L. Davis, D. A. Dixon, E. B. Garner, J. C. Gordon, M. H. Matus, B. Scott and F. H. Stephens, Angew. Chem., Int. Ed. Engl., 2009, 48, 6812–6816 CrossRef CAS PubMed.
- G. Chen, L. N. Zakharov, M. E. Bowden, A. J. Karkamkar, S. M. Whittemore, E. B. Garner, T. C. Mikulas, D. A. Dixon, T. Autrey and S.-Y. Liu, J. Am. Chem. Soc., 2014, 137, 134–137 CrossRef PubMed.
- C. D. Dimitrakopoulos and D. J. Mascaro, IBM J. Res. Dev., 2001, 45, 11–27 CrossRef CAS.
- Z. J. Zhou, X. P. Li, Z. B. Liu, Z. R. Li, X. R. Huang and C. C. Sun, J. Phys. Chem. A, 2011, 115, 1418–1422 CrossRef CAS PubMed.
-
(a) C.-W. Chen, M.-H. Lee and S. J. Clark, Nanotechnology, 2004, 15, 1837–1843 CrossRef CAS;
(b) R. H. Baughman, A. A. Zakhidov and W. A. d. Heer, Science, 2002, 297, 787–792 CrossRef CAS PubMed.
-
(a) A. H. Castro Neto, N. M. R. Peres, K. S. Novoselov and A. K. Geim, Rev. Mod. Phys., 2009, 81, 109–162 CrossRef CAS;
(b) Y. W. Son, M. L. Cohen and S. G. Louie, Nature, 2006, 444, 347–349 CrossRef CAS PubMed;
(c) S. Dutta, A. Manna and S. Pati, Phys. Rev. Lett., 2009, 102, 096601 CrossRef.
- Y. Bai, Z. J. Zhou, J. J. Wang, Y. Li, D. Wu, W. Chen, Z. R. Li and C. C. Sun, J. Mol. Model., 2013, 19, 3983–3991 CrossRef CAS PubMed.
- H. Q. Wu, R. L. Zhong, Y. H. Kan, S. L. Sun, M. Zhang, H. L. Xu and Z. M. Su, J. Comput. Chem., 2013, 34, 952–957 CrossRef CAS PubMed.
- R. F. W. Bader, Atoms in molecules: A Quantum Theory, Oxford University Press, Oxford, 1990 Search PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Wallingford CT, Gaussian, Inc., 2013 Search PubMed.
- K. Kawaguchi, J. Chem. Phys., 1992, 96, 3411–3415 CrossRef CAS PubMed.
- H. Fujimoto, S. Kato, S. Yamabe and K. Fukui, J. Chem. Phys., 1974, 60, 572–578 CrossRef CAS PubMed.
-
(a) W. Chen, Z. R. Li, D. Wu, Y. Li and C. C. Sun, J. Chem. Phys., 2005, 123, 164306 CrossRef PubMed;
(b) M. Iwaoka, H. Komatsu, T. Katsuda and S. Tomoda, J. Am. Chem. Soc., 2004, 126, 5309–5317 CrossRef CAS PubMed.
- H. L. Schmider and A. D. Becke, J. Mol. Struct., 2000, 527, 51–61 CrossRef CAS.
- A. Savin, O. Jepsen, J. Flad, O. K. Andersen, H. Preuss and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1992, 31, 187–188 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra10156e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.