
Green and all-carbon asymmetric supercapacitor based on polyaniline nanotubes and anthraquinone functionalized porous nitrogen-doped carbon nanotubes with high energy storage performance†
Ning An,
Yufeng An,
Zhongai Hu*,
Yadi Zhang,
Yuying Yang and
Ziqiang Lei
Key Laboratory of Eco-Environment-Related Polymer Materials of Ministry of Education, Key Laboratory of Polymer Materials of Gansu Province, College of Chemistry and Chemical Engineering, Northwest Normal University, Lanzhou, Gansu 730070, China. E-mail: zhongai@nwnu.edu.cn; Fax: +86 931 8859764; Tel: +86 931 7973255
First published on 14th July 2015
Abstract
Aqueous electrolyte-based asymmetric supercapacitors (ASCs) are one of the hot topics in the field of energy storage due to their high ionic conductivity, their environmental friendliness and lower cost. However, most research work on ASCs has involved non-renewable metal oxides (or hydroxides). Herein, an all-carbon and high-energy asymmetric supercapacitor (ASC) is constructed using polyaniline nanotubes (PNTs) as the positive electrode and anthraquinone-functionalized porous nitrogen-doped carbon nanotubes (AQ@PNCNTs) as the negative electrode. The PNTs are prepared by a facile chemical self-assembly method, and further carbonization/activation of the PNT precursor results in the formation of the porous nitrogen-doped carbon nanotubes (PNCNTs). Under solvothermal conditions, PNCNTs serve as a conductive substrate to adsorb anthraquinone (AQ) molecules, which can contribute additional electrochemical capacitance to the overall capacitance of the electrode. The as-assembled AQ@PNCNTs//PNTs ASC exhibits excellent supercapacitive performances in 1 M H2SO4 aqueous electrolyte. In particular, the device can deliver an energy density as high as 32.7 W h kg−1 at a power density of 700 W kg−1. Even at the power density of 14.0 kW kg−1, the energy density still remains at 20.2 W h kg−1. This strategy provides a feasible way to construct green supercapacitors with high power density and energy density.
1. Introduction
With the fast development of society and the economy, the whole world is facing the emerging ecological concern of fossil fuel exhaustion. Environmentally friendly, efficient and low-cost energy conversion and storage systems will play an important role in solving the intractable problem of energy sources. Supercapacitors, also called electrochemical capacitors, have been extensively used in many areas such as back-up power sources, hybrid electric vehicles, aeronautical facilities, and energy storage for wind energy due to their high power density, outstanding cycling stability and improved safety.1–3 However, the large-scale industrialization of supercapacitors remains restricted by their relatively low energy density.4 According to the equation E = 0.5CV2, high energy density (E) can be achieved by maximizing the specific capacitance (C) and the operating voltage (V) in practical devices. For this purpose, asymmetric supercapacitors (ASCs) are usually fabricated using two different electrode materials to enlarge the cell voltage.5–7Among various electrode materials for ASCs, transition-metal oxides (such as RuO2,8 MnO2,9 NiO10 and Co3O4 11 etc.) have been extensively investigated since they can provide multiple oxidation states for Faradaic reactions. However, most transition-metal oxides show a limited reversibility and rate capability during the charge/discharge process because of the transformation of crystalline phases along with volume expansion/shrinkage. More importantly, transition-metals are a non-renewable natural resource, and there has been increasing awareness of the depletion of transition-metal reserves. To meet the requirement of “green energy storage devices”, recently intense attention has been focused on environmentally friendly electrode materials such as conducting polymers and polymer-based carbon materials.12–14 Among these conducting polymers, polyaniline (PANI), which has emerged as a series of nanostructures (e.g. PANI nanofiber,15 nanotube16 and nanowire,17 and the growth of PANI on various porous carbon materials18), shows both high capacitive and low-cost advantages. To match the electrochemical behaviors of the PANI electrode, carbonaceous materials are commonly used as the negative electrode in the ASCs due to their excellent electrochemical stability in the negative potential range. Unfortunately, their limited capacitance values necessitate heavy use of carbonaceous materials to balance the relatively high capacitance of the PANI positive electrode. Consequently, the cell specific capacitance (or the energy density) is inevitably limited when carbonaceous materials are directly used in these capacitors.19 A new strategy is to try to attach environmentally friendly organic molecules with redox activity to carbonaceous materials. Such organic molecules can store and deliver charges by reversible Faradaic reactions on the surface of a conductive substrate, which contributes electrochemical capacitance to the overall capacitance of the electrodes. Importantly, most of these organic molecules are usually involved in multielectron Faradaic reactions, resulting in greater charge storage ability beyond most transition-metal oxides.20 Moreover, organic molecules derived from special organic functional groups can provide the desired electrochemical properties for the electrode materials. So this field has attracted intense research interest, even if the investigation and knowledge of it are in their infancy. For example, Pickup and Bélanger et al. improved the specific capacitance through either anthraquinone (AQ) modified carbon fabric or active carbon.21–24
In the present work, firstly we synthesized PNTs by a facile chemical self-assembly method, which exhibit a high supercapacitive performance (414 F g−1 at 1 A g−1) in the potential region of −0.2 to 1.0 V. Secondly, PNCNTs were prepared via a simple carbonization/activation route by using PNTs as the precursor. In order to enhance capacitive performance of the resultant materials within the low potential region, AQ as the functionalizing molecule was anchored on PNCNTs through a solvothermal process. As a result, the as-prepared AQ@PNCNTs exhibit a specific capacitance of 448 F g−1 in the potential window of −0.4 to 0.4 V, which are well able to match the PNTs for ASCs. Finally, an all-carbon and high-energy ASC device was successfully constructed by integrating the PNTs (positive electrode) and the AQ@PNCNTs (negative electrode). The AQ@PNCNTs//PNTs ASC device shows a maximum energy density of 32.7 W h kg−1 based on the total mass of two electrodes.
2. Experimental
2.1 Materials
Aniline monomer (An, Shanghai Zhongqin Chemical Reagent Corp, China) was distilled under reduced pressure. Anthraquinone (AQ, Alfa-Aesar, American), citric acid monohydrate (CA, Sinopharm Chemical Reagent Corp, China), and ammonium persulfate (APS, Sinopharm Chemical Reagent Corp, China) were used as received. All the experiments were carried out using deionized (DI) water and analytical reagents.2.2 Sample preparation
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 followed by evaporation at 80 °C under vacuum. The dried mixture was again heated at 800 °C under N2 atmosphere for 1.5 h. After cooling down to room temperature, the as-prepared products were washed with dilute HCl solution (1 M) and DI water several times until reaching neutral pH, and then dried overnight at 80 °C in a vacuum drying oven. Secondly, PNCNTs were functionalized by AQ molecules through a solvothermal process. Briefly, AQ (0.05 g) was dissolved in 60 mL dimethylformamide (DMF) by sonication and then PNCNTs (0.1 g) were dispersed and stirred in the above solution at room temperature until a black homogeneous dispersion was formed. Finally, the mixture was transferred into an 80 mL Teflon-lined stainless steel autoclave. After heating at 180 °C for 12 h, the dark sample was washed with DI water several times and then dried at 80 °C in a vacuum drying oven for 12 h to obtain the AQ@PNCNTs composite.
:3 followed by evaporation at 80 °C under vacuum. The dried mixture was again heated at 800 °C under N2 atmosphere for 1.5 h. After cooling down to room temperature, the as-prepared products were washed with dilute HCl solution (1 M) and DI water several times until reaching neutral pH, and then dried overnight at 80 °C in a vacuum drying oven. Secondly, PNCNTs were functionalized by AQ molecules through a solvothermal process. Briefly, AQ (0.05 g) was dissolved in 60 mL dimethylformamide (DMF) by sonication and then PNCNTs (0.1 g) were dispersed and stirred in the above solution at room temperature until a black homogeneous dispersion was formed. Finally, the mixture was transferred into an 80 mL Teflon-lined stainless steel autoclave. After heating at 180 °C for 12 h, the dark sample was washed with DI water several times and then dried at 80 °C in a vacuum drying oven for 12 h to obtain the AQ@PNCNTs composite.2.3 Material characterization
The morphologies of the samples were observed by field-emission scanning (FESEM; ULTRA Plus, Germany) and transmission (TEM; JEM-2010, Japan) electron microscopes. Fourier-transform infrared (FT-IR) spectra were analyzed by a Nicolet Nexus 670 Fourier transform infrared spectrometer. Powder X-ray diffraction (XRD) patterns were recorded on a diffractometer (D/Max-2400, Japan) equipped with Cu-Kα radiation (λ = 1.5418 Å). A Brunauer–Emmett–Teller (BET) measurement was conducted using a Micromeritics ASAP 2020 nitrogen adsorption apparatus (USA). Chemical state analysis of samples was performed by X-ray photoelectron spectroscopy (XPS; Escalab 210 system, Germany) with a monochromatic Al-Kα radiation source (ThermoVG Scientific).2.4 Electrochemical measurements
3. Results and discussion
3.1 Physical characterization
The morphologies of the as-synthesized electrode materials were imaged by FESEM and TEM. Fig. 1a and b show the typical FESEM images of PNTs. From the images, it can be observed that the PNTs are very long and present a highly entangled network structure. The high magnification image (Fig. 1b) illustrates that there are many PANI nanoparticles uniformly growing on the external surface of these tubes. Fig. 1c and d show the TEM images of PNTs at different magnifications. It can be seen that the PNTs exhibit an obvious tubular structure and that they are intertwined with each other, which is in agreement with the results of SEM. The FESEM image of AQ@PNCNTs (Fig. 1e) reveals that, on the whole, AQ@PNCNTs still maintain a one-dimensional tubular structure with a slightly rougher surface. To further characterize the structure of AQ@PNCNTs, TEM studies were carried out (Fig. 1f). Observation at high magnification (inset of Fig. 1f) shows that the average outer diameter of the hollow structure is about 120 to 150 nm, and that the tube wall thickness is around 50 nm. This unique structure not only supplies sufficient electrochemically active sites on the surface of PNCNTs but also serves as a micro-pool to store electrolyte, which can shorten the transport path of ions and facilitate the Faradaic reaction of AQ. It should be noted that, from the FESEM and TEM images of AQ@PNCNTs, there were no anthraquinone crystals, indicating that AQ was adsorbed on the PNCNTs’ surface in the form of the molecule. Compared with PNTs, AQ@PNCNTs show similar morphology after carbonization and functionalizing processes. We speculate it is mainly related to the following factors: firstly, the firm tubular structure with the thick tube wall (ca. 50 nm) and slow heating rate (5 °C min−1) prevent structural collapse during the carbonization process; secondly, through the non-covalent functionalization, most of the AQ molecules are anchored on the PNCNTs’ surface in the form of the molecule rather than the crystal, which has little impact on the morphology of the samples. | ||
| Fig. 1 (a and b) FESEM and (c and d) TEM images of the as-synthesized PNTs at different magnifications; (e) FESEM image of the AQ@PNCNTs; (f) the TEM image of AQ@PNCNTs exhibits a nanotube morphology (high magnification inset in subfigure). | ||
Typical nitrogen isotherm adsorption–desorption measurements of samples are shown in Fig. 2a and b. According to the IUPAC classification, the PNTs exhibit a characteristic type-V isotherm with a specific surface area of 23.96 m2 g−1. Moreover, from the N2 adsorption–desorption isotherms of PNTs, we can conclude the existence of macropores, corresponding to the small adsorbed volume. As shown in Fig. 2b, N2 adsorption–desorption isotherms of PNCNTs and AQ@PNCNTs have a typical type-IV hysteresis loop. The sharp increase of the adsorption amount at a low relative pressure P/P0 (ca. 0.01) implies that a large number of micropores were generated due to the gas release (e.g., CO, CO2, H2O) during the pyrolysis of the PNTs.26 In the range of P/P0 = 0.2–0.9, an almost horizontal plateau may be contributed to the adsorption in mesopores. A significant capillary condensation step and continuous increase of N2 absorption near the maximum relative pressure (at 0.9 to 0.99 P/P0) indicate the existence of a certain amount of macropores (exceeding 50 nm in diameter) into the samples.27 It should be noted that the specific surface areas of PNCNTs and AQ@PNCNTs are 1753.6 and 1005.5 m2 g−1, respectively. Namely, through non-covalent functionalization, some of the micropores in the PNCNTs are covered by AQ molecules. The detailed specific surface area and pore structure parameters of samples are summarized in Table S1 (ESI†).
 | ||
| Fig. 2 N2 adsorption–desorption isotherms of PNTs (a), PNCNTs and AQ@PNCNTs (b); XRD patterns (c) and FT-IR spectra (d) of samples. | ||
The XRD patterns of PNTs and AQ@PNCNTs are shown in Fig. 2c. Obviously, the PNTs have two primary characteristic peaks at ca. 20.7 and 24.6°, which are attributed to the periodic parallel and perpendicular scattering of the PANI chains respectively. At the same time, the result indicates that polymer nanotubes are partly crystalline owing to their tubular structures.28,29 The XRD pattern of AQ@PNCNTs exhibits two broad diffraction bands centered at 2θ = 23.0 and 43.1°, which are attributable to the (002) and (101) reflections of hexagonal carbon material (JCPDS PDF no. 75-1621), respectively.30 In addition, there is a weak diffraction peak at about 10.9°, which is another characteristic peak of CNTs.31 The FT-IR spectra of the samples are given in Fig. 2d. The FT-IR spectrum of the PNTs is consistent with the reported results in the literature.12 For instance, the presence of two noticeable peaks at 1573 cm−1 and 1500 cm−1 are owing to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching deformation of quinoid and benzene rings, respectively. The peaks at 1306 cm−1 and 1246 cm−1 correspond to the C–N and CN stretching vibrations of an aromatic amine. And the aromatic C–H in-plane bending and the out of plane deformation in the 1,4-disubstituted benzene ring are located at 1141 cm−1 and 804 cm−1.32 As shown in Fig. 2d, the characteristic peaks of AQ@PNCNTs at 1672, 1588 and 1285 cm−1 correspond to the CO stretching vibrations, aromatic CC vibrations and C–O skeletal vibration respectively, which are in accordance with the FT-IR spectrum of pure AQ (Fig. S1, ESI†). Besides, the bands of typical aromatic C–H vibrations are revealed at 693, 805 cm−1.33 These results strongly confirm that AQ molecules have functionalized the carbon substrate by π–π stacking interaction.
C stretching deformation of quinoid and benzene rings, respectively. The peaks at 1306 cm−1 and 1246 cm−1 correspond to the C–N and CN stretching vibrations of an aromatic amine. And the aromatic C–H in-plane bending and the out of plane deformation in the 1,4-disubstituted benzene ring are located at 1141 cm−1 and 804 cm−1.32 As shown in Fig. 2d, the characteristic peaks of AQ@PNCNTs at 1672, 1588 and 1285 cm−1 correspond to the CO stretching vibrations, aromatic CC vibrations and C–O skeletal vibration respectively, which are in accordance with the FT-IR spectrum of pure AQ (Fig. S1, ESI†). Besides, the bands of typical aromatic C–H vibrations are revealed at 693, 805 cm−1.33 These results strongly confirm that AQ molecules have functionalized the carbon substrate by π–π stacking interaction.
XPS is an effective method to characterize the surface chemical composition and chemical state of carbonaceous materials. As shown in Fig. 3a, the XPS spectrum of AQ@PNCNTs demonstrates a principal C 1s peak (∼284.8 eV), an O 1s peak (∼535 eV), and a pronounced N 1s (∼400 eV) without evidence of impurities, which confirms the successful incorporation of N atoms into the PNCNTs by the carbonization process.34 In the high resolution scan (Fig. 3b), the N 1s spectrum can be fitted by four peaks located at ca. 398.3 eV (pyridine-N), 399.9 eV (pyrrol-N), 401.1 eV (quaternary-N) and 403.1 eV (pyridine-N-oxide), respectively.35 As has been reported many times in the literature, carbon materials containing N atoms not only considerably contribute to improve wettability but also increase the number of chemically reactive sites and improve the holistic specific capacitance of PNCNTs.14 As shown in Fig. 3c, the C 1s XPS spectrum of AQ@PNCNTs is composed of five different binding states: C–C (∼284.8 eV), C–N (∼285.2 eV), C–O (∼286.4 eV), CO (∼287.7 eV) and O–CO (∼289.1 eV).36
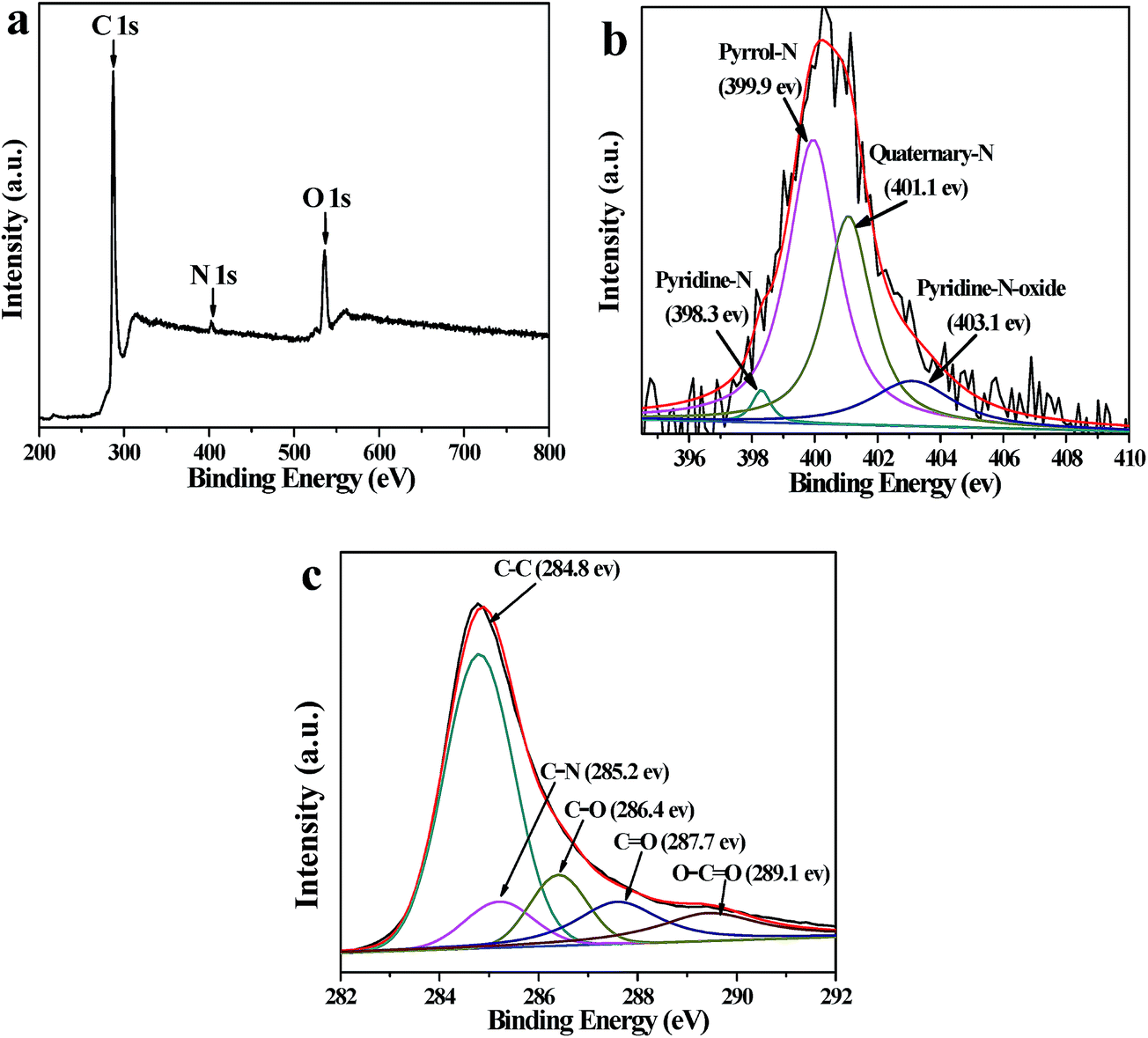 | ||
| Fig. 3 XRS spectra: (a) survey scan, and the (b) N 1s and (c) C 1s regions for AQ@PNCNTs. | ||
3.2 Electrochemical measurements
The electrochemical performances of AQ@PNCNTs and PNTs were analyzed using CV and GCD measurements in the three-electrode system. Fig. 4a and b show the CV curves of AQ@PNCNTs (negative electrode) and PNTs (positive electrode) at different scan rates, respectively. The excellent reversibility of the electrochemical behavior is displayed in the well-defined and symmetrical redox peaks of the AQ@PNCNTs CV curves. Due to the uncompensated resistance, the anodic and cathodic peaks move to higher and lower potentials respectively with increasing the scan rate. At a scan rate of 50 mV s−1, the peak separation is up to 69.5 mV. Additionally the ipa/ipc ratio is nearer 1.0, indicating that this redox process is reversible and faster. The peak separation (Eps), and peak current ratio (ipa/ipc) of the AQ@PNCNTs are collected in Table S2 (ESI†). We speculate that the three-dimensional highly entangled network structure of PNCNTs can increase the effective liquid–solid interfacial area, supply sufficient electrochemically active sites, and shorten the diffusion distance for the electrolyte ions. Consequently, the conversion of anthraquinone to 9,10-dihydroxyanthracene occurs at a relatively low charge-transfer resistance and exhibits the features of a fast dynamic process. The corresponding two-electron two-proton electrochemical process is described as follows:37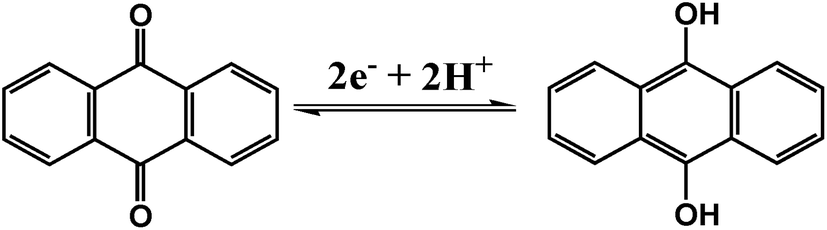
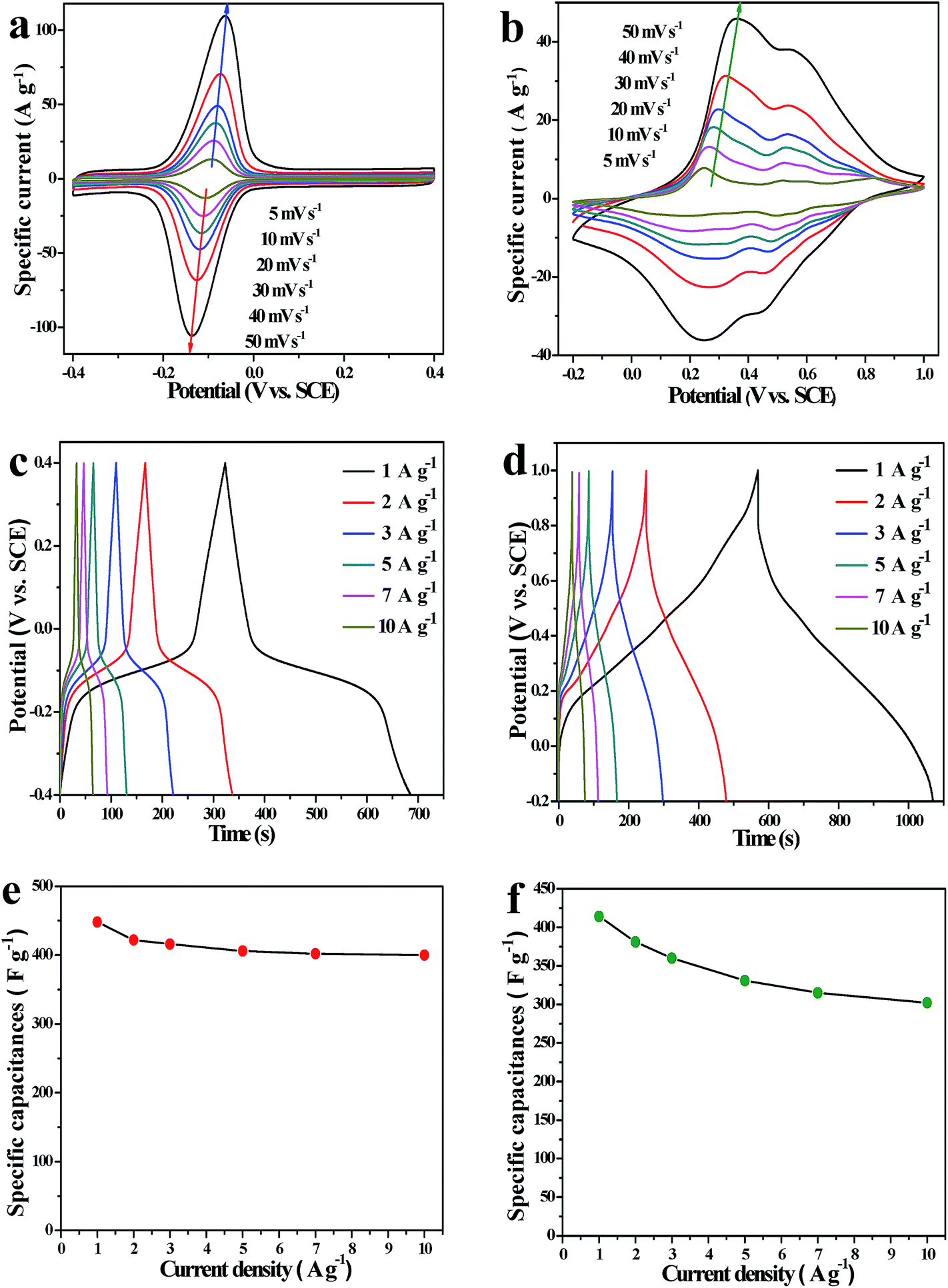 | ||
| Fig. 4 Electrochemical performances of the as-synthesized electrode materials in the three-electrode system: (a and b) CV curves of AQ@PNCNTs and PNTs with various scan rates in 1 M H2SO4 electrolyte, respectively; (c and d) GCD curves of AQ@PNCNTs and PNTs at different current densities, respectively; (e and f) specific capacitances of AQ@PNCNTs and PNTs at different current densities, respectively. | ||
As shown in Fig. 4b, all the CV curves of the PNTs exhibit a similar shape, and the characteristic CV shapes are not substantially impacted on increasing the scan rate from 5 to 50 mV s−1. This suggests that PNTs have a good electrochemical reversibility because of the fast diffusion of electrolyte ions into the tubular structure. The CV curve shows four couples of PANI typical redox peaks at 5 mV s−1 in the range from −0.2 to 1.0 V (Fig. S2, ESI†). The first peaks at around 0.24 V/0.18 V are ascribed to the PANI redox conversion between semi-conductive leucoemeraldine and conducting polaronicemeraldine. The second peaks at about 0.52/0.49 V are due to the formation and reduction of other structures (e.g. the p-benzoquinone and hydroquinone couple). The last two couples of redox peaks (at 0.59/0.57 V and 0.83/0.79 V) are related to the redox transition of p-aminophenol/benzoquinone imine and bipolaronic pernigraniline/protonated quinonediimine, respectively.38
To exactly assess the charge storage ability of AQ@PNCNTs and PNTs, the GCD test at different current densities was also performed in 1 M H2SO4. All the charge curves are almost symmetric to the corresponding discharging curves in Fig. 4c and d, evidencing a fast and highly reversible electrochemistry reaction. The relationship between current density and specific capacitance is illustrated in Fig. 4e and f. The specific capacitance of AQ@PNCNTs is as high as 448 F g−1 at 1 A g−1. It is worth mentioning that the specific capacitance of AQ@PNCNTs can still be maintained at 89% with the current density increasing from 1 to 10 A g−1. This high rate capability is probably related to the synergistic effect between the PNCNTs and AQ molecules, and the unique structure of the composite. First, the three-dimensional porous network structure can increase the effective liquid–solid interfacial area or the electrochemically active sites, and shorten the diffusion pathway length for the ions during the charging/discharging process. Secondly, the PNCNTs as an excellent conductive substrate enable the improvement of the overall conductivity of the composite because they can provide continuous charge transfer pathways. Finally, AQ molecules are anchored on the surface of PNCNTs and introduce a fast Faradaic reaction. As the positive electrode material, the specific capacitance of the PNTs is calculated to be 414 F g−1 at 1 A g−1, and is still retained at 302 F g−1 with the current density increased to 10 A g−1 (Fig. 4f). The PNTs also exhibit a high rate capability that may be due to the entangled tubular structures, which offer more ion channels and short diffusion paths to facilitate ion collection and fast transportation.
As shown in Fig. 5a, the CV curve exhibits a pair of reversible redox peaks on the basis of EDLC, which arises completely from the electrochemical capacitance of AQ molecules. According to the formula given in the ESI†, we use electrochemical techniques to quantitatively determine the AQ content in AQ@PNCNTs. In Fig. 5a, Va and Vb (V) are the selected boundaries of the potential range and the integrated area of the region (S1 or S2) is numerically equal to the charge contributed by the cathodic or anodic reaction of AQ.39 The calculations show that the average AQ content is about 27.1% of the total mass.
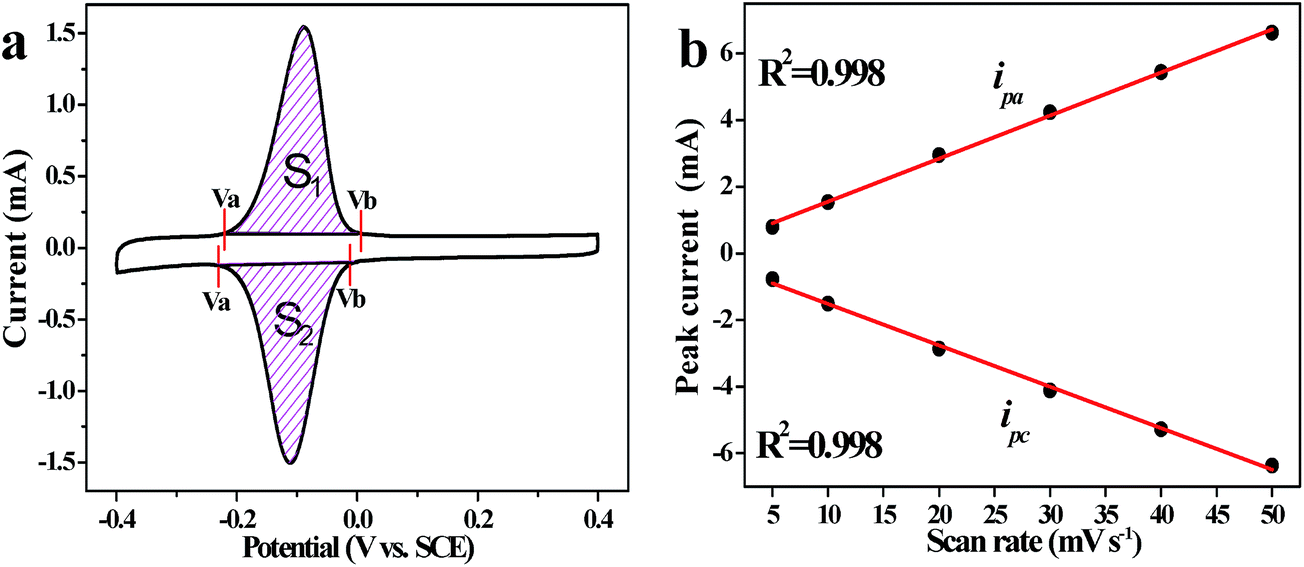 | ||
| Fig. 5 (a) CV curves of AQ@PNCNTs at 10 mV s−1 in 1 M H2SO4; (b) the relationship between the peak current (i) of AQ@PNCNTs and the scan rate. | ||
The relationship between the current peak (i) and the applied sweep rate (v) will determine whether the redox reaction is diffusion-controlled or surface-controlled (capacitive). For a redox reaction limited by semi-infinite diffusion, i varies as v1/2; for surface-controlled, it varies directly as v.40 From Fig. 5b, a linear relationship is detected between the current response and v with R2 = 0.998. Besides, the peak voltage differences in the CV curves of AQ@PNCNTs are small and almost independent of increasing scan rates, indicating this electrode process is limited neither by electrochemical polarization nor concentration diffusion. So the electrode process of AQ@PNCNTs is similar to a capacitive process.
3.3 Performances of the AQ@PNCNTs//PNTs ASC
For practical applications, the ASC device was assembled based on AQ@PNCNTs as the negative electrode and PNTs as the positive electrode using 1 M H2SO4 as the electrolyte. According to the potential window for AQ@PNCNTs (−0.4 to 0.4 V) and PNTs (−0.2 to 1.0 V), it is expected that the cell voltage of the AQ@PNCNTs//PNTs ASC can be extended to 1.4 V (Fig. 6a). To achieve a satisfactory electrochemical performance, the charge balance should follow the relationship q+ = q−, where the charge stored is dependant on the specific capacitance (C), the potential range in the GCD process (ΔE), and the mass of the electrode (m) following the equation: q = C × ΔE × m.41 Thus, the optimal loading mass ratio between PNTs and AQ@PNCNTs was estimated to be m(PNTs)/m(AQ@PNCNTs) = 0.72. | ||
| Fig. 6 (a) CV curves of the individual electrodes, tested in the three-electrode system at 10 mV s−1; (b) CV curves of the AQ@PNCNTs//PNTs ASC at different scan rates in 1 M H2SO4 electrolyte; (c) GCD curves and (d) specific capacitance of the ASC at various specific currents; (e) cycling stability of the AQ@PNCNTs//PNTs ASC; (f) schematic illustration of the as-assembled ASC device and a picture showing that two tandem ASC devices can light up a red LED indicator (2.0 V). | ||
The CV curves for the ASC devices at various scan rates show that the cell voltage of the AQ@PNCNTs//PNTs capacitor was up to 1.4 V in 1 M H2SO4. Furthermore, a pair of redox peaks (at ca. 0.5 V) is attributed to the reversible Faradaic reaction of the electrode materials observed in Fig. 6b. Fig. 6c shows the GCD curves of the AQ@PNCNTs//PNTs ASC at different current densities from 1 A g−1 to 20 A g−1, in which a pair of obvious voltage plateaux at low potentials is in agreement with the CV curves. Additionally, all charging curves are almost symmetric to their corresponding discharging curves, indicating a good Coulombic efficiency and an excellent electrochemical reversibility. The specific capacitance of the AQ@PNCNTs//PNTs ASC is calculated based on the total mass of the two electrodes. As shown in Fig. 6d, the specific capacitance is up to 120 F g−1 at 1 A g−1 and still retains 74 F g−1 at 20 A g−1, which is superior to most values previously reported for other ASCs, such as Co3O4@MnO2//MEGO (49.8 F g−1 at 1 A g−1),6 NiO/GF//HPNCNT (116 F g−1 at 1 A g−1),13 rGO/CNT/PPy//Mn3O4 (40 F g−1 at 1 A g−1).42 The long-term cycling stability of the supercapacitor was measured by repeating the GCD test in the range from 0 to 1.4 V at a current density of 5 A g−1 for 1000 cycles (Fig. 6e). During the first 200 cycles, the specific capacitance of the AQ@PNCNTs//PNTs ASC slightly increases with cycle number, implying that the full release of the electrochemical capacitance needs an activated process. After activation, the capacitance retention is 80% over the next 800 cycles, indicating a good long-term stability of the ASC. In order to demonstrate the practicability of the AQ@PNCNTs//PNTs ASC, a light-emitting diode (LED) with a working voltage of 2.0 V was powered by the ASC. After being charged to 2.0 V, the tandem ASCs (two supercapacitors connected in series) could easily light up a red LED (Fig. 6f).
Fig. 7a shows a Ragone plot relative to the corresponding energy and power densities of the AQ@PNCNTs//PNTs ASC. Notably, the ASC device can deliver a maximum energy density (Emax) of 32.7 W h kg−1 at a power density of 700 W kg−1 and remained 20.2 W h kg−1 at 14.0 kW kg−1. This is much higher than recently reported ASCs in aqueous electrolyte, such as graphene/PANI//graphene/RuO2 (26.3 W h kg−1),8 C-AQ//RuO2 (26.7 W h kg−1),22 C-AQ//C-DHB (10.0 W h kg−1).24 Table 1 summarizes the reported ASCs devices constructed from various electrodes. A radar plot was drafted as shown in Fig. 7b to overall assess the electrochemical performance of the as-assembled AQ@PNCNTs//PNTs ASC. Additionally, from the EIS analysis of AQ@PNCNTs//PNTs ASC (Fig. S3†), a negligible resistor–capacitor loop in the high-frequency region and an almost vertical spike in the low-frequency region were observed, indicating a low charge-transfer resistance (Rct) at the electrode/electrolyte interface and an obvious capacitive behavior with a small diffusion resistance.47
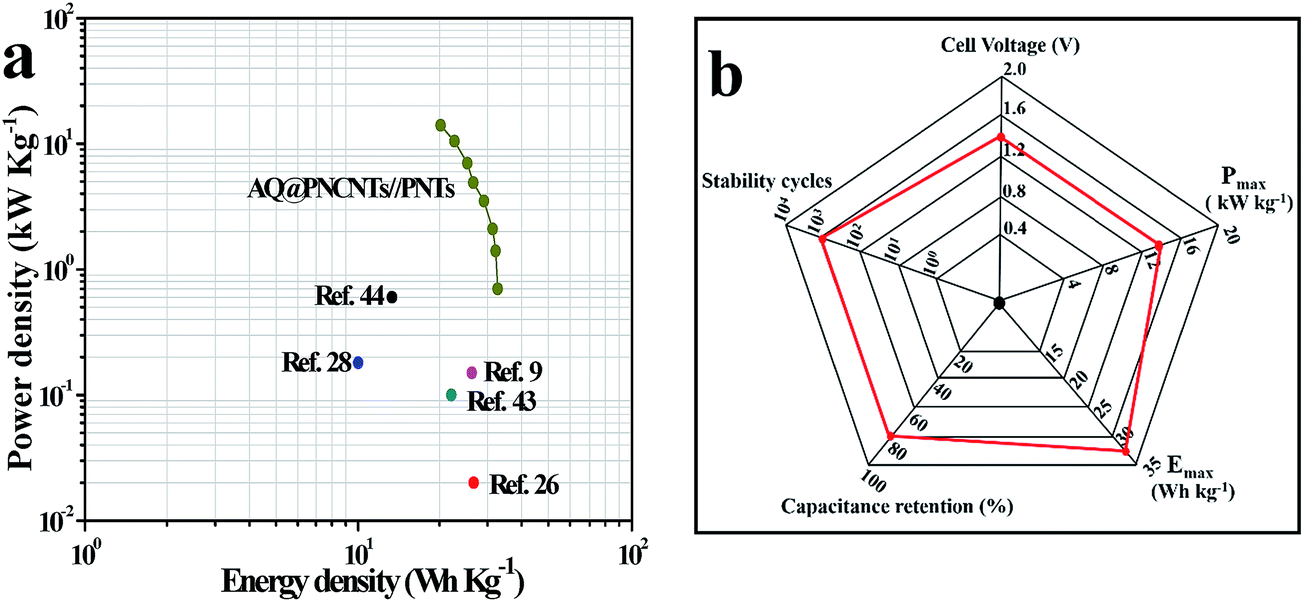 | ||
| Fig. 7 (a) Ragone plot of the AQ@PNCNTs//PNTs ASC in comparison to the ASCs recently reported in the literature; (b) radar plots of the ASC. The red curve is generated by connecting data points from the AQ@PNCNTs//PNTs ASC using 1 M H2SO4 electrolyte. | ||
| Asymmetric cell | Electrolyte | Vcell (V) | Ccell (F g−1) | Emax (W h kg−1) | Pmax (kW kg−1) | Ref. |
|---|---|---|---|---|---|---|
| AQ@PNCNTs//PNTs | H2SO4 | 1.4 | 120 | 32.7 | 14.0 | Present work |
| CNT/MnO2//CNT/In2O3 | Na2SO4 | 2.0 | 46 | 25.5 | 50.3 | 7 |
| RGO–RuO2//RGO–PANI | KOH | 1.4 | 100 | 26.3 | 49.8 | 8 |
| C-AQ//RuO2 | H2SO4 | 1.3 | 109 | 26.7 | 10.8 | 22 |
| C-AQ//C-DHB | H2SO4 | 1.2 | 63 | 10.0 | 6.3 | 24 |
| rGO/CNT/PPy//Mn3O4 | NaNO3 | 1.6 | 74 | 14.3 | 6.62 | 42 |
| Co3O4@MnO2//MEGO | LiOH | 1.6 | 49.8 | 17.7 | 158 | 43 |
| NiO//porous carbon | KOH | 1.5 | 38 | 10.1 | 10 | 6 |
| IL-CNT–graphene gel//MnO2–graphene gel | Na2SO4 | 1.8 | 57 | 25.6 | 20.5 | 44 |
| CoO@PPy nanowire//AC | Na2SO4 | 1.6 | 67.5 | 21.4 | 2.1 | 45 |
| CoMoO4@3D graphene//AC | KOH | 1.8 | 85 | 21.1 | 0.6 | 46 |
4. Conclusions
In summary, a novel all-carbon negative electrode material (AQ@PNCNTs) was designed, and fabricated on the basis of multiple steps, for supercapacitors. AQ as the functionalizing molecule is anchored on the PNCNTs through π–π stacking interactions, which is essential to enhance the supercapacitive performance of the resultant electrode materials in the negative potential region. The PNCNT cores are used as a conductive network to facilitate electron collection and fast transport during the electrochemical reaction. The as-assembled AQ@PNCNTs//PNTs ASC can achieve a high energy density in aqueous solution.Acknowledgements
The authors gratefully acknowledge the financial support offered by the National Natural Science Foundation of China (20963009 and 21163017) and Specialized Research Fund for the Doctoral Program of Higher Education (No. 20126203110001).References
- D. Vonlanthen, P. Lazarev, K. A. See, F. Wudl and A. J. Heeger, Adv. Mater., 2014, 23, 5095–5100 CrossRef PubMed.
- H. Chen, L. F. Hu, M. Chen, Y. Yan and L. M. Wu, Adv. Funct. Mater., 2014, 24, 934–942 CrossRef CAS PubMed.
- H. W. Wang, Z. A. Hu, Y. Q. Chang, Y. L. Chen, H. Y. Wu, Z. Y. Zhang and Y. Y. Yang, J. Mater. Chem., 2011, 21, 10504–10511 RSC.
- Q. T. Qu, S. B. Yang and X. L. Feng, Adv. Mater., 2011, 23, 5574–5580 CrossRef CAS PubMed.
- Z. Li, Z. W. Xu, H. L. Wang, J. Ding, B. Zahiri, C. M. B. Holt, X. H. Tan and D. Mitlin, Energy Environ. Sci., 2014, 7, 1708–1718 CAS.
- M. Huang, Y. X. Zhang, F. Li, L. L. Zhang, Z. Y. Wen and Q. Liu, J. Power Sources, 2014, 252, 98–106 CrossRef CAS PubMed.
- P. C. Chen, G. Z. Shen, Y. Shi, H. Chen and C. W. Zhou, ACS Nano, 2010, 4, 4403–4411 CrossRef CAS PubMed.
- J. T. Zhang, J. W. Jiang, H. L. Li and X. S. Zhao, Energy Environ. Sci., 2011, 4, 4009–4015 CAS.
- L. Li, Z. A. Hu, N. An, Y. Y. Yang and H. Y. Wu, J. Phys. Chem. C, 2014, 118, 22865–22872 CAS.
- S. I. Kim, J. S. Lee, H. J. Ahn, H. K. Song and J. H. Jang, ACS Appl. Mater. Interfaces, 2013, 5, 1596–1603 CAS.
- X. H. Xia, J. P. Tu, Y. J. Mai, X. L. Wang, C. D. Gu and X. B. Zhao, J. Mater. Chem., 2011, 21, 9319–9325 RSC.
- J. J. Mu, G. F. Ma, H. Peng, J. J. Li, K. J. Sun and Z. Q. Lei, J. Power Sources, 2013, 242, 797–802 CrossRef CAS PubMed.
- H. Wang, H. Yi, X. Chen and X. F. Wang, J. Mater. Chem. A, 2014, 2, 3223–3230 CAS.
- H. Peng, G. F. Ma, K. J. Sun, J. J. Mu, M. T. Luo and Z. Q. Lei, Electrochim. Acta, 2014, 147, 54–61 CrossRef CAS PubMed.
- H. Y. Mi, X. G. Zhang, S. D. Yang, X. G. Ye and J. M. Luo, Mater. Chem. Phys., 2008, 112, 127–131 CrossRef CAS PubMed.
- W. Chen, R. B. Rakhi and H. N. Alshareef, J. Mater. Chem. A, 2013, 1, 3315–3324 CAS.
- K. Wang, Q. H. Meng, Y. J. Zhang, Z. X. Wei and M. H. Miao, Adv. Mater., 2013, 25, 1494–1498 CrossRef CAS PubMed.
- Z. Q. Niu, P. S. Luan, Q. Shao, H. B. Dong, J. Z. Li, J. Chen, D. Zhao, L. Cai, W. Y. Zhou, X. D. Chen and S. A. Xie, Energy Environ. Sci., 2012, 5, 8726–8733 CAS.
- H. L. Wang, Q. M. Gao and J. Hu, J. Power Sources, 2010, 195, 3017–3024 CrossRef CAS PubMed.
- T. Tomai, S. Mitani, D. Komatsu, Y. Kawaguchi and I. Honma, Sci. Rep., 2014, 4, 3591–3596 Search PubMed.
- K. Kalinathan, D. P. DesRoches, X. Liu and P. G. Pickup, J. Power Sources, 2008, 181, 182–185 CrossRef CAS PubMed.
- Z. Algharaibeh, X. Liu and P. G. Pickup, J. Power Sources, 2009, 187, 640–643 CrossRef CAS PubMed.
- G. Pognon, T. Brousse, L. Demarconnay and D. Bélanger, J. Power Sources, 2011, 196, 4117–4122 CrossRef CAS PubMed.
- Z. Algharaibeh and P. G. Pickup, Electrochem. Commun., 2011, 13, 147–149 CrossRef CAS PubMed.
- S. Giri, D. Ghosh and C. K. Das, Adv. Funct. Mater., 2014, 24, 1312–1324 CrossRef CAS PubMed.
- W. T. Huang, H. Zhang, Y. Q. Huang, W. K. Wang and S. C. Wei, Carbon, 2011, 49, 838–843 CrossRef CAS PubMed.
- X. X. Xiang, Z. Z. Huang, E. Liu, H. J. Shen, Y. Y. Tian, H. Xie, Y. H. Wu and H. Z. Wu, Electrochim. Acta, 2011, 56, 9350–9356 CrossRef CAS PubMed.
- L. Y. Zhang and M. X. Wan, Adv. Funct. Mater., 2013, 13, 815–820 CrossRef PubMed.
- J. P. Pouget, M. E. Jozefowicz, A. E. A. Epstein, X. Tang and A. G. MacDiarmid, Macromolecules, 1991, 24, 779–789 CrossRef CAS.
- F. Su, C. K. Poh, J. S. Chen, G. Xu, D. Wang, Q. Li and X. W. Lou, Energy Environ. Sci., 2011, 4, 717–724 CAS.
- F. Liu, S. Y. Song, D. F. Xue and H. J. Zhang, Adv. Mater., 2012, 24, 1089–1094 CrossRef CAS PubMed.
- L. J. Zhang, Y. Z. Long, Z. J. Chen and M. X. Wan, Adv. Funct. Mater., 2004, 14, 693–698 CrossRef CAS PubMed.
- M. Büschel, C. Stadler, C. Lambert, M. Beck and J. Daub, J. Electroanal. Chem., 2000, 484, 24–32 CrossRef.
- Y. Zhao, C. G. Hu, Y. Hu, H. H. Cheng, G. Q. Shi and L. T. Qu, Angew. Chem., 2012, 124, 11533–11537 CrossRef PubMed.
- D. Hulicova-Jurcakova, M. Kodama, S. Shiraishi, H. Hatori, Z. H. Zhu and G. Q. Lu, Adv. Funct. Mater., 2009, 19, 1800–1809 CrossRef CAS PubMed.
- Y. Li, Y. Zhao, H. H. Cheng, Y. Hu, G. Q. Shi, L. M. Dai and L. T. Qu, J. Am. Chem. Soc., 2011, 134, 15–18 CrossRef PubMed.
- M. Quan, D. Sanchez, M. F. Wasylkiw and D. K. Smith, J. Am. Chem. Soc., 2007, 129, 12847–12856 CrossRef CAS PubMed.
- H. Peng, G. F. Ma, J. J. Mu, K. J. Sun and Z. Q. Lei, J. Mater. Chem. A, 2014, 2, 10384–10388 CAS.
- N. An, F. H. Zhang, Z. A. Hu, Z. M. Li, L. Li, Y. Y. Yang and Z. Q. Lei, RSC Adv., 2015, 5, 23942–23951 RSC.
- P. Simon, Y. Gogotsi and B. Dunn, Science, 2014, 343, 1210–1211 CrossRef CAS PubMed.
- J. Yan, Z. J. Fan, W. Sun, G. Q. Ning, T. Wei, Q. Zhang, R. F. Zhang, L. J. Zhi and F. Wei, Adv. Funct. Mater., 2012, 22, 2632–2641 CrossRef CAS PubMed.
- Y. J. Peng, T. H. Wu, C. T. Hsu, S. M. Li, M. G. Chen and C. C. Hu, J. Power Sources, 2014, 272, 970–978 CrossRef CAS PubMed.
- D. W. Wang, F. Li and H. M. Cheng, J. Power Sources, 2008, 185, 1563–1568 CrossRef CAS PubMed.
- Y. Sun, Y. Cheng and K. He, RSC Adv., 2015, 5, 10178–10186 RSC.
- L. Q. Fan, G. J. Liu, J. H. Wu, L. Liu, J. M. Lin and Y. L. Wei, Electrochim. Acta, 2014, 137, 26–33 CrossRef CAS PubMed.
- X. Z. Yu, B. A. Lu and Z. Xu, Adv. Mater., 2014, 26, 1044–1051 CrossRef CAS PubMed.
- J. G. Wang, Y. Yang, Z. H. Huang and F. Kang, Carbon, 2013, 61, 190–199 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09943a |
| This journal is © The Royal Society of Chemistry 2015 |