DOI:
10.1039/C5RA09919F
(Paper)
RSC Adv., 2015,
5, 76689-76695
Electrochemical investigation of the stable chromium species in molten FLINAK
Received
26th May 2015
, Accepted 3rd September 2015
First published on 4th September 2015
Abstract
The electrochemical behavior of Cr(III) in FLINAK melts at 600 °C was studied through cyclic voltammetry (CV) and square wave voltammetry (SWV). Cr(III) reduction proceeded in two steps: initial Cr(III) reduction to Cr(II) followed by subsequent reduction to Cr. The peak corresponding to the reduction of Cr(III) to Cr(II) was also observed in SWVs and CVs even though only CrF2 was added to the FLINAK melts. This result confirms that Cr(II) was converted to Cr(III) via disproportion: 3Cr(II) = 2Cr(III) + Cr. Such a conversion could be attributed to the formation of the stable species [CrF6]3−, as determined through Raman spectroscopy.
1. Introduction
Molten fluorides are suitable for use as coolants of molten salt reactors because these materials present excellent heat transfer properties.1–3 However, pyrohydrolysis of the absorbed water left in raw molten fluorides commonly occurs upon melting, and formation of HF is inevitable [eqn (1)]. Given its high solubility in fluorides,4,5 HF (g) remains in the salts and causes corrosion of structural materials.6–9 This corrosion is caused by the reaction of Cr and HF,10,11 as shown in eqn (2). Despite this problem, however, high-temperature Cr-bearing alloys, such as Hastelloy, are still used in molten fluorides because of their acceptable, time-tested, and overall high-temperature properties.12,13| | |
H2O + 2F− ↔ O2− + 2HF (H2O + F− ↔ OH− + HF)
| (1) |
| | |
Cr + xHF ↔ (x/2)H2 + CrFx
| (2) |
The Oak Ridge National Laboratory (ORNL) performed periodic sampling and off-line chemical analysis and found that the corrosion valence state of Cr is determined by the acid–base properties of the molten fluoride employed;14 here, a Lewis acid is defined as a fluoride ion acceptor, and a Lewis base is a fluoride ion donor. In LiF–NaF–ZrF4, for example, the Cr(III)/Cr(II) ratio increases with increasing basicity of the melt (i.e., decreases in the amount of ZrF4).15 Pure FLINAK consisting of LiF, NaF, and KF [LiF![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :NaF:KF, 46.5:11.5:42 (mol%)] is known to be a strongly basic solvent that tends to stabilize the Cr(III) valence state.14 Using the theory of non-electric transfer reported by Ozeryanaya16 as a basis, Olson17 speculated that the Cr in FLINAK salt dissolves as a form of Cr(II), which would be further converted to Cr(III). Yoko et al.15,18,19 proposed that disproportionation of 3Cr(II) → 2Cr(III) + Cr probably occurs. Unfortunately, no relevant studies have yet been published to prove this disproportionation.
:NaF:KF, 46.5:11.5:42 (mol%)] is known to be a strongly basic solvent that tends to stabilize the Cr(III) valence state.14 Using the theory of non-electric transfer reported by Ozeryanaya16 as a basis, Olson17 speculated that the Cr in FLINAK salt dissolves as a form of Cr(II), which would be further converted to Cr(III). Yoko et al.15,18,19 proposed that disproportionation of 3Cr(II) → 2Cr(III) + Cr probably occurs. Unfortunately, no relevant studies have yet been published to prove this disproportionation.
Nourry20 recently found that U(III) could be oxidized to U(IV) in LiF–CaF2–UF3 melts through cyclic voltammetry (CV) and square wave voltammetry (SWV). Bing21 proposed that Fe3+ could be converted to Fe2+ in KCl–CaCl2–NaCl–MgCl2 melts by CV. Manning22 also used CV to verify that O2− and O22− are stable in LiF–BeF2–ZrF4 and LiF–BeF2–ThF4 melts whereas O2− is unstable in these eutectics. Reports thus far20–24 suggest that the electrochemical method can be used to investigate the stable valence of electro-active species because the technique offers quick responses and in situ monitoring.
The aim of the present work is to determine the stable valence state of Cr in FLINAK melts through CV and SWV. Further study of the stable species of FLINAK–CrF3 and FLINAK–CrF2 was performed through Raman spectroscopy.
2. Experimental
Highly-purified (99.98 wt%) LiF–NaF–KF [46.5:11.5:42 (mol%)] [FLINAK] eutectic salt was supplied by Shanghai Institute of Organic Chemistry, Chinese Academy of Sciences. The Cr(III) concentration varying from 58 to 1500 ppm was added into FLINAK melts in the form of CrF3 (Sigma-Aldrich, 99.99%), while Cr(II) varying from 600 to 1300 ppm was added in the form of CrF2 (Alfa Aesar, 99%). The fluorides were melted at 600 °C in a vitreous carbon crucible placed in a stainless steel cell inside an electric furnace. The temperature was measured by a nickel–chromium thermocouple positioned just outside the crucible.
During electrochemical measurement, a platinum wire (1.0 mm diameter) was used as the working electrode; the surface area of this wire was determined by measuring the immersion depth in the melts after completion of the experiment. The auxiliary electrode was a graphite rod (6.0 mm diameter) with a large surface area (2.5 cm2). All potentials were referenced to a platinum wire (1.0 mm diameter) that had been proven to function as a quasi-reference electrode Pt/PtOx/O2−, with a stable potential when the O2− concentration is constant.25,26 CV and SWV were performed on a computer-controlled AUTOLAB digital electrochemical analyzer (Metrohm AutoLab Co., Ltd). The entire electrochemical assembly was placed inside a glove box, as shown in Fig. 1, and the typical concentrations of moisture and oxygen in the glove box were generally below 2 ppm.
 |
| | Fig. 1 Drawing of the experimental cell (1): glove box. (2): Transition cover. (3): Water cooling coil let. (4): Alumina insulation tubes. (5): Working electrode. (6): Reference electrode. (7): Thermocouple. (8): Auxiliary electrode. (9): Vitreous carbon crucible and molten salt. (10): Furnace. | |
Raman analysis was performed to identify the structures of salt. The HR 800 (HORIBA Jobin Yvon) Raman system equipped with CCD detector was used in the triple configuration. The 532 nm line of an argon ion laser with 200 mW average power was used for exciting the sample. Techniques of back scattering and confocal collection were employed to collect laser signal, which would be focused on the inlet slit (200 μm width) of a monochromator (800 mm focal distance) and eventually collected by CCD detector after passing through the outlet slit of the monochromator. The system was interfaced with a personal computer and the spectra were saved in digital form. We carried out in situ studies using a Linkam TS1000 environmental cell. The heating unit consists of a ceramic furnace having a sealed chamber that allows vacuuming and purging to be performed. With an aim to avoid of deliquesce, the FLINAK sample was initially loaded into a platinum crucible in glove box, and then quickly transferred to the furnace with an S-type Pt/Rh thermocouple in intimate contact. The sample was dried under vacuum for more than 2 h at 200 °C, followed by heating to 600 °C and keeping it for about one hour under argon atmosphere. Afterwards, the laser was focused on the sample through a silica window in the cover of the cell equipped with an independent water cooling system.
3. Results and discussion
3.1. Electrochemical behavior of Cr(III) in FLINAK melts
3.1.1. Cyclic voltammetry. A typical cyclic voltammogram of the FLINAK melts after addition of 308 ppm Cr(III) was obtained on a platinum electrode at 600 °C and is shown in Fig. 2. Besides one couple of cathodic/anodic signals D and D′ corresponding to the deposition and dissolution of alkali metal, two reduction peaks, A and B, at −0.65 and −0.87 V (vs. Pt), respectively, in the cathodic run and two anodic counter-peaks, A′ and B′, at −0.54 and −0.66 V (vs. Pt), respectively, can be observed. Since the deposition potential of Cr is more positive than that of alkali metal in a fluoride system, the peaks of A and B were attributed to the reduction of Cr(III).
 |
| | Fig. 2 Cyclic voltammograms of FLINAK–Cr(III) (308 ppm) melts at 600 °C and scan rate of 0.2 V s−1. Working El.: Pt (S = 1.13 cm2); auxiliary El.: graphite; reference El.: Pt. | |
 |
| | Fig. 3 Cyclic voltammograms of FLINAK–Cr(III) (308 ppm) melts at different scan rate from 0.05 to 0.4 V s−1 at 600 °C. Working El.: Pt (S = 1.13 cm2); auxiliary El.: graphite; reference El.: Pt. | |
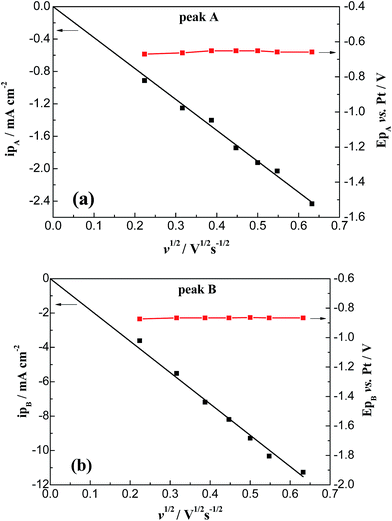 |
| | Fig. 4 Plots of peak current density and peak potential vs. the square root of the scan rate in FLINAK–Cr(III) (308 ppm) melts at 600 °C for cathodic peaks A and B. Working El.: Pt (S = 1.13 cm2); auxiliary El.: graphite; reference El.: Pt. | |
3.1.2. Square wave voltammetry. The use of SWV for analytical purposes, which is theoretically valid for reversible systems, can be extended to other systems as long as the criterion of linearity between the peak intensity and the square root of the frequency signal is observed.33–37 The typical square wave voltammograms of FLINAK melts after addition of 308 ppm Cr(III) obtained on a platinum electrode at 10–45 Hz are shown in Fig. 5(a). Two reduction peaks, A and B, at about −0.55, and −0.83 V (vs. Pt), respectively, can be observed. And the peak current densities of δipA and δipB show a linear relationship with the square root of the frequency in the range of 10–45 Hz, as shown in Fig. 5(b) and (c). By measuring the half peak width, W1/2, the number of exchanged electrons can be calculated by the following equation:38,39| |
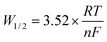 | (3) |
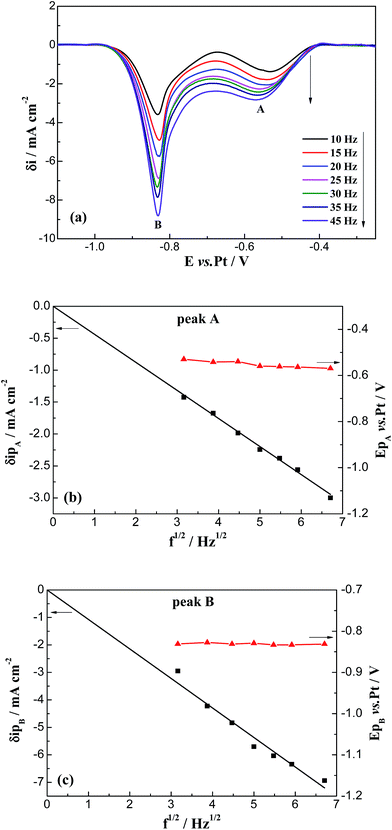 |
| | Fig. 5 (a) Square wave voltammograms of FLINAK–Cr(III) (308 ppm) melts at 600 °C. (b and c) Plots of peak current intensity and peak potential vs. the square root of the frequency for cathodic peaks A and B. Frequency signal: 10–45 Hz, pulse height: 20 mV, step potential: 2 mV. Working El.: Pt (S = 1.13 cm2); auxiliary El.: graphite; reference El.: Pt. | |
The electron transfer numbers of peaks A and B are calculated as 1 and 2, respectively. Thus, peak A at −0.55 V (vs. Pt) and peak B at −0.83 V (vs. Pt) correspond to the reductions of Cr(III)/Cr(II) and Cr(II)/Cr, respectively. These results are in accordance with those obtained through CV.
At a given temperature of 600 °C, from the obtained SWVs, the current density of the reduction peak for Cr(III)/Cr(II) have a linear relation with Cr(III) concentration at a signal frequency of 10 Hz, as showed in Fig. 6. The equation of this straight line is:
| | |
δipA (mA cm−2) = −0.00493[Cr(III)] (ppm)
| (4) |
 |
| | Fig. 6 Linear relationship between peak current intensity (δipA) and Cr(III) concentration in FLINAK melts at 600 °C. Frequency signal: 10 Hz, pulse height: 20 mV, step potential: 2 mV. Working El.: Pt (S = 1.13 cm2); auxiliary El.: graphite; reference El.: Pt. | |
This equation can be used to determine the Cr(III) concentration in molten FLINAK.
3.2. Electrochemical behavior of Cr(II) in FLINAK melts
Fig. 7 shows the square wave voltammograms of FLINAK–Cr(II) (1300 ppm) melts on a platinum electrode at 600 °C and different holding times. Peaks A and B, which are assigned to the reductions of Cr(III)/Cr(II) and Cr(II)/Cr, respectively, can be observed even though only CrF2 was added to the FLINAK melts. Besides, the A/A′ attributed to Cr(III)/Cr(II) redox system also can be observed in FLINAK–CrF2 melts by cyclic voltammetry, as shown in Fig. 8. Thus, both SWV and CV results proved that the Cr(II) will be converted to Cr(III), which is more stable in FLINAK melts. Moreover, through scanning electron microscope-energy dispersive spectrometry (SEM-EDS) and X-ray diffraction (XRD) analysis, the deposited metal Cr can be found at the bottom surface of solidified FLINAK–CrF2 salt, as shown in Fig. 9(a) and (b). These observations indicate that Cr(II) can be converted to Cr(III) and metal Cr through disproportionate reaction as shown in eqn (5):| | |
3Cr(II) = 2Cr(III) + Cr
| (5) |
 |
| | Fig. 7 Square wave voltammograms of FLINAK–Cr(II) (1300 ppm) melts recorded on a platinum electrode at 600 °C and different holding times. Pulse height: 20 mV, step potential: 2 mV, frequency: 10 Hz. Working El.: Pt (S = 1.13 cm2); auxiliary El.: graphite; reference El.: Pt. | |
 |
| | Fig. 8 Cyclic voltammograms of FLINAK–Cr(II) (1300 ppm) melts recorded on a platinum electrode at 600 °C and different holding times. Scan rate: 0.1 V s−1. Working El.: Pt (S = 1.13 cm2); auxiliary El.: graphite; reference El.: Pt. | |
 |
| | Fig. 9 Analysis of the bottom surface of solidified FLINAK–CrF2 salt by (a) SEM-EDS and (b) XRD techniques. | |
3.3. Conversion ratio of Cr(II) to Cr(III)
Plots of the cathodic peak current density (δipA and δipB) of FLINAK–Cr(II) (1300 ppm) melts versus holding time at 600 °C are shown in Fig. 10; here, δipA increases from −1.92 mA cm−2 to −3.58 mA cm−2 and then reaches a plateau at about −3.75 mA cm−2 after 140 min. δipB increases from −3.25 mA cm−2 to −5.09 mA cm−2 within the first 120 min and then suddenly decreases to −4.50 mA cm−2 at 140 min. This sudden decrease at 140 min suggests that Cr(II) is nearly depleted in the system because of disproportionation. After 140 min, the concentrations of Cr(II) and Cr(III) remain nearly constant due to the equilibrium of disproportionation. Thus, the δipA and δipB assigned to the reduction current densities of Cr(III)/Cr(II) and Cr(II)/Cr eventually remain at −3.75 and −4.40 mA cm−2, respectively.
 |
| | Fig. 10 Plots of peak current density (δipA and δipB) vs. holding time at 600 °C as measured from the SWVs of FLINAK–Cr(II) (1300 ppm) melts. Frequency signal: 10 Hz, pulse height: 20 mV, step potential: 2 mV. Working El.: Pt (S = 1.13 cm2); auxiliary El.: graphite; reference El.: Pt. | |
As it is stated in the eqn (4) that δipA has a linearity with Cr(III) concentration, when δipA equaled to −3.75 mA cm−2, the concentration of Cr(III) was found to be 761 ppm. Meanwhile, based on the disproportion reaction shown in eqn (5), 761 ppm Cr(III) was converted from 1141 ppm Cr(II). That means, the conversion ratio of Cr(II) to Cr(III) was approximately equal to 87.77% since the initial concentration of Cr(II) was 1300 ppm, as shown in eqn (6).
| |
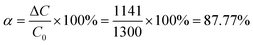 | (6) |
where
α is the conversion ratio of Cr(
II) to Cr(
III),
C0 is the initial concentration of Cr(
II) and Δ
C is Cr(
II) involved in disproportionate reaction.
When the added concentration of Cr(II) varied from 600 to 1300 ppm, the conversion ratio of Cr(II) to Cr(III) is 86.71–88.23% estimated from the SWV analysis within the experimental error, as shown in Table 1.
Table 1 Independence of conversion ratio on Cr(II) concentration
| [Cr(II)]/ppm |
δipA/mA cm−2 |
Conversion ratio (α) |
| 600 |
−1.71 |
86.71% |
| 900 |
−2.61 |
88.23% |
| 1300 |
−3.75 |
87.77% |
3.4. Determination of structures of Cr(III) and Cr(II) in FLINAK melts through Raman spectroscopy
Yoko18 speculated that the stable complex ion [CrF6]3− may be present in FLINAK melts. Using first-principles molecular dynamics modeling, Nam40 also predicted the presence of [CrF6]3− in molten FLINAK. To verify this speculation, the FLINAK–CrF3 and FLINAK–CrF2 melts were extracted on-line by using a nickel sampler at 600 °C and rapidly quenched. Then c.a. 0.1 g samples were transferred to a platinum crucible for Raman analysis. The predicted Raman spectra of [CrF6]3− was calculated according to density functional theory (DFT) using the (U) B3LYP method with the LanL2DZ basis set. No imaginary frequency modes were observed at the stationary states of the optimized structures.
Fig. 11 shows the Raman spectra obtained in FLINAK–Cr(III) fluorides. No detectable peak can be observed in pure FLINAK [Fig. 11(a)], which indicates that completely free ions exist in the systems studied. When 2000 ppm Cr(III) was added to FLINAK, two peaks, located at 269 and 561 cm−1, were observed at 200 °C as shown in Fig. 11(b). As the test temperature increased to 600 °C, the sample melted and two peaks at 274 and 545 cm−1 were also observed in Fig. 11(c), which agreed well with that measured at 200 °C. These peaks may be ascribed to the Cr(III) complex. In order to validate this structure, the predicted Raman spectrum of [CrF6]3− was determined through DFT calculation and the result was shown in Fig. 11(d). Two main peaks at about 274 and 554 cm−1, which were assigned to Cr–F cutting and Cr–F breathing vibrations, respectively, can be observed, which is in a good agreement with that reported by J. Mi41 and L. Daimay.42 The good agreement between the experimental and calculated results indicate that the stable structure of Cr(III) in molten FLINAK is the [CrF6]3− complex.
 |
| | Fig. 11 Raman spectra of the pure FLINAK and FLINAK–Cr(III) (2000 ppm) sample. (a) FLINAK sample, (b) FLINAK–Cr(III) (2000 ppm) sample measured at 200 °C, (c) FLINAK–Cr(III) (2000 ppm) sample measured at 600 °C, (d) the calculated [CrF6]3− complex. | |
FLINAK melts obtained after addition of 1300 ppm Cr(II) were also subjected to Raman analysis and the results were shown in Fig. 12. Two main peaks assigned to the Cr(III) complex can be observed at both 200 and 600 °C, as shown in Fig. 12(a) and (b), indicating that Cr(III) with the [CrF6]3− structure was more stable in molten FLINAK–CrF2 system. This result supports our conclusion from the SWV and CV results that Cr(II) is converted to Cr(III) with the [CrF6]3− structure in molten FLINAK. It should be noted that several weak signals appear at about 334, 418 and 486 cm−1 as shown in Fig. 12(b). These peaks may be attributed to residual Cr(II) that had not been completely converted to Cr(III).
 |
| | Fig. 12 Raman spectra of the FLINAK–Cr(II) (1300 ppm) sample. (a) Measured at 200 °C and (b) measured at 600 °C. | |
4. Conclusions
The electrochemical behavior of Cr(III) in molten FLINAK at 600 °C was studied through CV and SWV. Results showed that reduction of Cr(III) proceeds through two steps: initial reduction of Cr(III) to Cr(II) at −0.65 V (vs. Pt) and subsequent reduction to Cr at −0.87 V (vs. Pt).
The peak corresponding to reduction of Cr(III) to Cr(II) also appeared in the SWVs and CVs even though only CrF2 was added to the FLINAK melts. Besides, the deposited Cr metal can be found at the bottom surface of solidified FLINAK–CrF2 salt. These results confirmed that Cr(II) was converted to Cr(III) via the disproportion: 3Cr(II) = 2Cr(III) + Cr. This disproportion equilibrium was established after CrF2 was added to FLINAK for 140 min and the final conversion ratio of Cr(II) to Cr(III) was in the range of 86.71–87.77% when initial concentration of Cr(II) varied from 600 to 1300 ppm. Such a conversion could be attributed to the generation of the more stable species [CrF6]3−, as identified through Raman spectroscopy.
Acknowledgements
This work was financially supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (Grant No. XDA02020400) and the Young Scientists Fund of the National Natural Science Foundation of China (Grant No. 51404237).
References
- M. Salanne, C. Simon, P. Turq and P. A. Madden, J. Fluorine Chem., 2009, 130, 38 CrossRef CAS PubMed.
- J. P. M. van der Meer and R. J. M. Konings, J. Nucl. Mater., 2007, 360, 16 CrossRef CAS PubMed.
- J. Křepel, B. Hombourger, C. Fiorina, K. Mikityuk, U. Rohde, S. Kliem and A. Pautz, Ann. Nucl. Energy, 2014, 64, 380 CrossRef PubMed.
- P. E. Field and J. H. Shaffer, J. Phys. Chem., 1967, 71, 3218 CrossRef CAS.
- J. H. Shaffer, W. R. Grimes and G. M. Watson, J. Phys. Chem., 1959, 63, 1999 CrossRef CAS.
- S. Delpech, C. Cabet, C. Slim and G. S. Picard, Mater. Today, 2010, 13, 34 CrossRef CAS.
- T. Nagasaka, M. Kondo, T. Muroga, N. Noda, A. Sagara, O. Motojima, A. Suzuki and T. Terai, J. Nucl. Mater., 2009, 386–388, 716 CrossRef CAS PubMed.
- M. Kondo, T. Nagasaka, A. Sagara, N. Noda, T. Muroga, Q. Xu, M. Nagura, A. Suzuki and T. Terai, J. Nucl. Mater., 2009, 386–388, 685 CrossRef CAS PubMed.
- M. Kondo, T. Nagasaka, Q. Xu, T. Muroga, A. Sagara, N. Noda, D. Ninomiya, M. Nagura, A. Suzuki, T. Terai and N. Fujii, Fusion Eng. Des., 2009, 84, 1081 CrossRef CAS PubMed.
- L. C. Olson, J. W. Ambrosek, K. Sridharan, M. H. Anderson and T. R. Allen, J. Fluorine Chem., 2009, 130, 67 CrossRef CAS PubMed.
- J. Qiu, Y. Zou, G. Yu, H. Liu, Y. Jia, Z. Li, P. Huai, X. Zhou and H. Xu, J. Fluorine Chem., 2014, 168, 69 CrossRef CAS PubMed.
- D. Williams, D. Wilson, J. Keiser, L. Toth and J. Caja, Research on Molten Fluorides as High Temperature Heat Transfer Agents, American Nuclear Society Winter Meeting, 2003 Search PubMed.
- H. E. McCoy, R. L. Beatty, W. H. Cook, R. E. Gehlbach, C. R. Kennedy, J. W. Koger, A. P. Litman, C. E. Sessions and J. R. Weir, Nucl. Technol., 1970, 8, 156 CAS.
- D. Williams, L. Toth and K. Clarno, in ORNL/TM-2006/12, 2006.
- J. Redman, in ORNL-2626, 1959.
- I. N. Ozeryanaya, Met. Sci. Heat Treat., 1985, 27, 184 CrossRef.
- L. C. Olson, Materials corrosion in molten LiF–NaF–KF eutectic salt, University of Wisconsin-Madison, 2009 Search PubMed.
- T. Yoko and R. A. Bailey, J. Electrochem. Soc., 1984, 131, 2590 CrossRef CAS.
- G. W. Mellors and S. Senderoff, Applications of Fundamental Thermodynamics to Metallurgical Processes, Gordon and Breach Science Publishers, New York, 1967 Search PubMed.
- C. Nourry, P. Soucek, L. Massot, R. Malmbeck, P. Chamelot and J.-P. Glatz, J. Nucl. Mater., 2012, 430, 58 CrossRef CAS.
- B. Li, J. Lou, H. Wang and J. Yu, J. Rare Earths, 2011, 29, 96 Search PubMed.
- D. L. Manning, J. Electrochem. Soc., 1977, 124, 480 CrossRef CAS PubMed.
- L. Cassayre, J. Serp, P. Soucek, R. Malmbeck, J. Rebizant and J.-P. Glatz, Electrochim. Acta, 2007, 52, 7432 CrossRef CAS PubMed.
- M. V. Smirnov and V. Y. Kudyakov, Electrochim. Acta, 1983, 28, 1339 CrossRef CAS.
- Y. Berghoute, A. Salmi and F. Lantelme, J. Electroanal. Chem., 1994, 365, 171 CrossRef CAS.
- A. D. Graves and D. Inman, Nature, 1965, 20, 481 CrossRef.
- A. J. Bard and R. L. Faulkner, Electrochemistry: Principles, Methods and Applications, Wiley, New York, 1980 Search PubMed.
- J. Wang, Analytical electrochemistry, Wiley, New York, 2000 Search PubMed.
- M. Straka, M. Korenko and F. Lisy, J. Radioanal. Nucl. Chem., 2010, 284, 245 CrossRef CAS.
- M. Gibilaro, L. Massot, P. Chamelot, L. Cassayre and P. Taxil, Electrochim. Acta, 2013, 95, 185 CrossRef CAS PubMed.
- R. A. Bailey, J. Appl. Electrochem., 1986, 16, 737 CrossRef CAS.
- D. Ludwig, L. Olson, K. Sridharan, M. Anderson and T. Allen, Corros. Eng., Sci. Technol., 2011, 46, 360 CrossRef CAS PubMed.
- P. Chamelot, L. Massot, L. Cassayre and P. Taxil, Electrochim. Acta, 2010, 55, 4758 CrossRef CAS PubMed.
- C. Hamel, P. Chamelot and P. Taxil, Electrochim. Acta, 2004, 49, 4467 CrossRef CAS PubMed.
- P. Chamelot, P. Palau, L. Massot, A. Savall and P. Taxil, Electrochim. Acta, 2002, 47, 3423 CrossRef CAS.
- P. Chamelot, B. Lafage and P. Taxil, Electrochim. Acta, 1997, 43, 607 CrossRef.
- L. Massot, L. Cassayre, P. Chamelot and P. Taxil, J. Electroanal. Chem., 2007, 606, 17 CrossRef CAS.
- L. Ramaley and M. S. Krause, Anal. Chem., 1969, 41, 1362 CrossRef CAS.
- L. Ramaley and M. S. Krause, Anal. Chem., 1969, 41, 1365 CrossRef.
- H. O. Nam, A. Bengtson, K. Vörtler, S. Saha, R. Sakidja and D. Morgan, J. Nucl. Mater., 2014, 449, 148 CrossRef CAS.
- J. Mi, S. Luo, H. Sun, X. Liu and Z. Wei, J. Solid State Chem., 2008, 181, 1723 CrossRef CAS PubMed.
- L. V. Daimay, B. C. Norman, G. F. William and G. G. Jeanette, The Handbook of Infrared and Raman Characteristic Frequencies of Organic Molecules, Academic Press, 1991 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 













