
Establishment of a long-term chick forebrain neuronal culture on a microelectrode array platform
Abstract
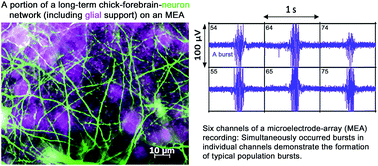
Biosensor systems formed by culturing primary animal neurons on microelectrode array (MEA) platforms are drawing increasing research interest for their potential as a rapid, sensitive and functional neurotoxicity assessment, as well as for many other electrophysiological related research purposes. In this paper we established a long-term chick forebrain neuron culture (C-FBN-C) on MEAs with a more than 5 month long lifespan and up to 5 month long stability in morphology and physiological functions; characterized C-FBN-C morphologically, functionally and developmentally; partially compared its functional features with its rodent counterpart; and discussed its pros and cons as a novel biosensor system in comparison to its rodent counterpart and human induced pluripotent stem cells (hiPSCs). Our results show that C-FBN-C on the MEA platform (1) can be used as a biosensor of its own type in a wide spectrum of basic biomedical research; (2) is of value in comparative physiology in cross-species studies; and (3) may have potential to be used as an alternative, cost-effective approach to its rodent counterpart within shared common functional domains (such as specific types of ligand-gated ion channel receptors and subtypes expressed in the cortical tissues of both species) in large-scale environmental neurotoxicant screening that would otherwise require millions of animals.
 Please wait while we load your content...
Please wait while we load your content...

