
Effect of the solvent on improving the recognition properties of surface molecularly imprinted polymers for precise separation of erythromycin†
Abstract
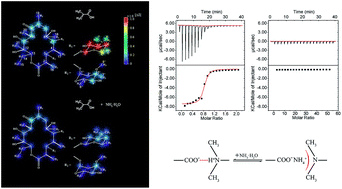
The formation of thin films with imprinting sites that can be accessed more rapidly by target molecules is an improvement in bulk molecular imprinting polymers. To achieve suitable properties for specific application, on the basis of our previous study of bulk erythromycin (ERY) imprinted polymers, we here rationally design, generate and test surface-imprinted polymers specific for ERY. ERY-A surface-imprinted glass fibers (GF-MIPs) were fabricated using glass fibers as the matrix, methacrylic acid (MAA) as the functional monomer, ethyleneglycol dimethacrylate (EGDMA) as the cross-linker and azobis(4-cyanovaleric acid) as the initiator. Then, we specifically studied the effect of the solvent on the recognition performance of the GF-MIPs. 13C-NMR and isothermal titration calorimetry (ITC) measurements were employed to demonstrate that the addition of NH3·H2O can block the interactions between the –COOH group and the tertiary amine on ERY-A. The results of static adsorption and dynamic separation experiments also indicate that an increase of specificity can easily be obtained by mediating the solvent environment, thus satisfying the more stringent requirements of precise separation.
 Please wait while we load your content...
Please wait while we load your content...

