
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Aqueous phase one-pot green synthesis of SnSe nanosheets in a protein matrix: negligible cytotoxicity and room temperature emission in the visible region†
Laboni
Das
,
Apurav
Guleria
and
Soumyakanti
Adhikari
*
Radiation & Photochemistry Division, Bhabha Atomic Research Centre, Mumbai 400 085, India. E-mail: asoumya@barc.gov.in; Fax: +91-22-25505331; Tel: +91-22-25590301
First published on 9th July 2015
Abstract
In recent times, SnSe has attracted considerable attention as an environmentally friendly alternative to cadmium- and lead-based photovoltaic materials. Herein, we report a rapid, facile, reproducible, and purely green method for the synthesis of SnSe nanosheets (NS) in an aqueous solution of bovine serum albumin (BSA). Interestingly, BSA itself acted as a reducing agent, stabilizing agent and shape directing template. Time dependent TEM imaging showed the self-assembly of the nanoparticles (NPs) into single crystalline NS. A probable mechanism has been proposed on the basis of the alteration of the intrinsic α-helix structure of BSA into β-pleated sheets at the pH (∼3.0) of the reaction mixture. This directs the self-assembly of small SnSe NPs into the NS like structure. It was further evident that the –NH and the –S–S– linkages in the protein are involved in the functionalization of the NPs. As grown SnSe nanomaterial was found to display room temperature photoluminescence (PL), which has been rarely reported. The PL spectra appeared to be the convolution of two bands at 425 and 470 nm, which were attributed to the band-gap and trap/surface state emission, respectively. Moreover, the reduction potential values obtained from the cyclic voltammetry indicate better thermodynamic feasibility for the reduction of SnSe, while the cytotoxicity studies revealed no toxic effects up to a concentration of 100 μM. This signifies the potential application of these nanomaterials in photovoltaics and biomedical contexts.
1. Introduction
During the last two decades, nanomaterials and quantum dots (QDs) of diverse metal chalcogenides such as CdSe, CdS, CdTe, ZnSe, and PbSe with varying morphologies have been prepared extensively owing to their tremendous optoelectronic applications.1–7 However, in recent times, the focus has shifted towards developing Cd and Pb free (due to their highly toxic nature) chalcogenide compounds.8 The quest for low toxic, cost-effective, and efficient photovoltaic materials continues to fuel new research in the field of semiconductor nanocrystal synthesis.Recently, tin(II) selenide (SnSe) has garnered interest as an environmentally benign material with potential applications in solar cells9,10 memory switching devices,11 holographic recording systems, infra-red, and other energy storage devices such as lithium ion batteries.12 The recent report by Zhao et al.13 illustrates ultralow thermal conductivity and high thermoelectric behavior of SnSe that makes it promising material for efficient conversion of waste heat into electrical energy. Basically, SnSe belongs to a family of IV–VI semiconductors and has an indirect band gap of 0.90 eV and a direct band gap of 1.30 eV in bulk phase.9 This reflects a good overlap of its band gap with that of the solar spectrum. SnSe has a layered GeS-type crystal structure that facilitates two-dimensional (2D) anisotropic growth.14 The research on 2D semiconductor nanostructures has also been the topic of recent interest. In spite of the impending role in future technologies, tin selenides has not received much attention compared to other chalcogenides. Therefore, SnSe being the newly emerging field of research interest, the integration of synthetic strategies with green chemistry would lead to greater dimensions to its applications.
Of course, significant amount of work have been reported on the novel synthesis of SnSe, but majority of these processes are very tedious, and involve stringent experimental conditions i.e. high temperature and toxic organic solvents like phosphines, oleylamine,15–19 use of electron beam radiation20 and laser induced reaction.21 The solid state route22 requires a CVD furnace setup for synthesis. However, one step, room temperature green synthesis of SnSe nanomaterials in aqueous medium has rarely been reported till date.
Herein, we have employed protein (bovine serum albumin, BSA) milieu as a soft template for the synthesis of SnSe nanosheets in aqueous solution. No external reducing agent was employed and the reaction was carried out at purely ambient conditions. The composition and the structural role of the BSA in directing the self-assembly of the initially produced nanoparticles into single crystalline nanosheets has been explained. The as grown SnSe nanosheets displayed room temperature PL, which was found to be the convolution of band gap as well as surface/trap state emission. The potential application of as grown nanomaterials in photovoltaics (Li-ion battery) was explored by carrying out the Cyclic Voltammetry (CV) studies. The reduction potential values obtained indicate better thermodynamic feasibility for the reduction of SnSe, which has been attributed to the intercalation of Li ions within the SnSe lattice. Apart from this, cytotoxicity studies of the nanomaterials was also carried out, which revealed negligible toxicity up to a concentration of 100 μM. The as employed protocol for the synthesis of SnSe nanomaterials aligns with the increasing emphasis on the topic of green chemistry, and further enhancing the possibility of unique and exciting applications of these nanomaterials, as discussed in the text.
2. Experimental
2.1. Chemicals
High purity chemicals i.e. tin(II) chloride dihydrate (SnCl2·2H2O), sodium sulfite (Na2SO3), selenium (Se) powder, bovine serum albumin (BSA) were obtained from Sigma-Aldrich and used as received. The reagents such as hydrochloric acid (HCl) and ethanol, used for preparation of Sn precursor and washing the precipitates were purchased with highest purity available from sd fine chemicals, Mumbai. Nanopure water (conductivity 0.06 μS cm−1) used in the preparation of precursor solutions and washing purposes was obtained from millipore water purifying system.2.2. Synthesis
The precursor solution of tin was prepared by drop wise addition of concentrated HCl to the aqueous solution of SnCl2·2H2O (10 mM), until the white precipitates of Sn(OH)Cl dissolves. The precursor solution of Sn was called as solution A. Essentially, SnCl2·2H2O forms an insoluble basic salt (Sn(OH)Cl) in aqueous solution. Therefore, to prepare a clear solution of SnCl2, HCl solution is added to maintain the equilibrium towards the left-hand of the eqn (1).| SnCl2(aq.) + H2O(l) = Sn(OH)Cl(s) + HCl(l) | (1) |
The procedure for preparing the stock precursor solution of Se i.e. sodium selenosulphate (Na2SeSO3) has been described elsewhere.23,24 The precursor solution of Se was named as solution B, and its concentration was adjusted to make it equivalent to that of solution A. In the preceding process, solution A was added to 30 μM BSA (prepared in nanopure water; pH ∼ 7.0) with continuous stirring and the resultant solution was labeled as C. On mixing solution B to solution C with continuous stirring, the color of the resulting solution turned to greenish yellow. In this procedure, equimolar concentration of both the precursors was used. This final solution was labeled as D. The precipitates for morphological characterization were obtained by centrifugation of the solution D at 9000 rotations per minute. The obtained precipitates were washed thrice with nanopure water followed by ethanol and acetone. Subsequently, the precipitates were dried under vacuum to obtain a yellow powder. It is to be emphasized here that no pre-requisites i.e. heating or inert atmosphere was involved in the above synthetic process.
2.3. Characterization
Optical absorption measurements were carried out by using a JASCO V-650 absorption spectrophotometer. Steady-state PL measurements were carried out at room temperature by using a Hitachi F-4500 spectrofluorimeter. X-ray diffraction (XRD) measurements were recorded on a Phillips X-ray diffractometer, model PW 1710 system, using a monochromatic Cu Kα source (λ = 1.54 Å). The instrument was well calibrated using standard samples before any measurements. The electron diffraction and transmission electron microscopic (TEM) images were acquired on a TEM, Philips model number CM200 with operating voltages 20–200 kV and resolution: 2.4 Å. HRTEM measurements were carried out on model number FEI, TECNAI-F30. Samples for TEM measurements were prepared by re-dispersing the as obtained precipitates in ethanol and depositing a drop of as prepared sol on thin carbon coated copper grid and the solvent was allowed to evaporate. Surface and morphological characterization of as obtained nanomaterials were carried out on JEOL JSM-T330 Scanning Electron Microscope (SEM). The FTIR spectra were recorded using a diamond single reflectance ATR probe in an IR Affinity-1 FTIR spectrometer. The X-ray photoelectron spectroscopy (XPS) of the nanomaterials was carried out using Mg Kα (1253.6 eV) source and a MAC-2 electron analyzer. The XPS-analysis chamber has a base vacuum of 10−9 mbar. The XPS binding energy scale was calibrated to C-1s line at 284.5 eV.PL lifetime measurements were carried out by using a time correlated single photon counting (TCSPC) instrument (model: IBH, UK). The instrument response function (IRF) of the setup was measured by collecting the scattered light from a TiO2 suspension in water. The samples were excited by a diode laser of output wavelength, 374 nm with an IRF of less than 200 ps.
The conformation changes in the protein structure were determined by circular dichroism (CD) spectroscopy using a BioLogic spectrometer (France, model no. MOS-500), fitted with a xenon lamp. A cell with a path length of 10 mm was used. Each spectrum was obtained after an average of three scans.
The cyclic voltammetric (CV) experiments were carried out using a potentiostat-galvanostat (Bio-Logic Science Instruments, France). A three-electrode cell was used, where a glassy carbon (diameter ∼ 3 mm) and a Pt wire served as the working electrode and counter electrode, respectively. Ag/AgCl, KCl (3 M) was used as the reference electrode. Tetraethyl ammonium tetrafluroborate (TEABF4) dissolved in 0.1 M acetonitrile was used as the supporting electrolyte. The as prepared nanomaterials were washed thoroughly with acetone before the experiments. 5 μl of the nanomaterials (re-dispersed in acetone) containing solution (concentration ∼ 13.6 mg ml−1) was drop-cast on the glassy carbon electrode to completely cover it up, and allowed to dry in vacuum oven for 10 minutes. Before the experiments, the electrolyte solution was continuously purged with high purity nitrogen for 15 minutes. In the experiments, the scan rate was fixed at 50 mV s−1.
Cytotoxicity studies were carried out in Chinese hamster ovary (CHO) cells, suspended in Dulbecco's modified Eagle's medium (DMEM) with 10% fetal calf serum seeded in 96 well plate at a density of 2 × 103 cells per well and incubated at 37 °C in 5% CO2 humidified atmosphere. After the cells attachment (12–16 hours), they were treated with increasing concentrations of test compounds for 24 hours, and cell viability was determined by a colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay as described previously.25 In brief, MTT solution (0.5 mg mL−1 in phosphate-buffered saline) was added to each well, and incubated for 4 hours at 37 °C. The blue formazan crystals formed were dissolved in solubilization buffer (10% SDS (sodium dodecyl sulphate) in 0.01 N HCl) and detected by absorbance measurement at 570 nm. Cell viability was expressed as percentage (%) viability compared to the control (100%).
3. Results and discussion
As already mentioned in the experimental section, the color of the resulting solution (on mixing precursor solutions) turned to greenish yellow. Fig. 1 illustrates the emergence of excitonic absorption at ∼350 nm, indicating the formation of tin selenide nanoparticles. The camera ready picture showing the greenish yellow solution of tin selenide nanoparticles has been provided in the inset of Fig. 1. | ||
| Fig. 1 UV-vis absorption spectra of tin selenide nanoparticles. Inset: picture showing the greenish yellow solution of as synthesized colloidal nanoparticles. | ||
The direct and indirect band gap values were determined from the Tauc plots (Fig. S1 & S2†) and obtained as 3.18 and 2.68 eV, respectively. Large blue shift (with respect to the bulk band gap values of 0.90 (indirect) and 1.30 eV (direct)) in the excitonic peak position clearly indicates that the as grown nanoparticles belong to the strong confinement regime. The direct band gap value of the nanoparticles obtained in the present work is comparable to the band gap value of 3.89 eV for the SnSe QDs synthesized by electron beam radiation, as reported by Li et al.20 Of late, Ling et al.26 also obtained large direct and indirect band gap values of 2.78 and 2.27 eV, respectively for the SnSe QDs synthesized by an interfacial chemical route.
To determine the chemical form of tin selenide (i.e. either SnSe or SnSe2), the nanoparticles were extracted in powder form (as discussed in the Experimental section) and characterization studies were conducted thereafter. The XRD of as obtained precipitates (picture shown in the inset of Fig. 2a) have been shown in Fig. 2a. The diffraction pattern of the nanoparticles was found to be in good agreement with the standard JCPDS card no. 32-1382, corresponding to the orthorhombic phase of SnSe. The diffraction peaks have been designated to their respective lattice planes (400) and (800).15,27,28 The sharp pointed peaks in XRD signify the highly crystalline nature of the nanomaterial. The HRTEM image has been provided in Fig. 2b, where lattice fringes could be clearly seen. The measured interplanar distance, d = 0.30 nm closely matches with the standard ‘d’ value of 0.305 nm for lattice plane (011) of orthorhombic phase of SnSe. In analogy with the XRD, XPS of the nanoparticles further substantiated the formation of SnSe. Fig. 2c represents the core level XPS spectra of Sn and Se. The 3d5/2 and 3d3/2 peaks for Sn appeared at 486.2 eV and 494.7 eV, respectively, which is an evidence of the presence of (+2) oxidation state of Sn in the nanomaterial. Moreover, a little higher binding energy of 486.5 eV and above corresponds to Sn(+4) state.29–31 The 3d5/2 and 3d3/2 peaks for Se appeared at 53.8 eV and 54.7 eV, respectively. These binding energy values for 3d electronic states of Sn and Se correspond to SnSe stoichiometry.32,33
 | ||
| Fig. 2 (a) XRD spectrum for SnSe nanomaterials corresponding to JCPDS card no. 32-1382. Inset shows the picture of SnSe nanomaterials extracted in the powder form; (b) HRTEM image showing the lattice fringe pattern of the nanomaterials; (c) core level XPS spectra of Sn 3d and Se 3d. | ||
To determine the binding sites of BSA involved in the interaction with the nanoparticles, FTIR spectra of pure BSA and BSA–SnSe were recorded (see Fig. 3). The IR signatures of pure BSA at 1246, 1391, 1520, 1630 and 3043 cm−1 are assigned to the C–N stretching/N–H bending vibrations of amide III, side chain COO− of the protein, N–H bending/C–H stretching vibrations of amide II, C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibrations of the peptide bond (belongs to amide I) and N–H stretching vibrations of amide A, respectively.34 The peak at 1630 cm−1 corresponds to short segment chains connecting α-helical segments of BSA.35 Also, a peak at ∼564 cm−1 was observed (in case of pure BSA), and could be attributed to the disulfide stretching vibrations.36 The strong resemblance of the FTIR spectra of pure BSA with the BSA–SnSe categorically indicates towards the significant coating of the former on the surface of the nanoparticles. Further, a red shift of ∼28 cm−1 and 46 cm−1 was observed in the disulphide peak (from 564 to 536 cm−1) and bending vibrations of amide III (from 1246 to 1200 cm−1), respectively in the FTIR spectrum of BSA–SnSe as compared to that of pure BSA. On the other hand, a blue shift of 27 cm−1 (from 3043 to 3070 cm−1) and 4 cm−1 (from 1520 to 1524 cm−1) was noticed in the N–H stretching vibrations of amide A and N–H bending vibration of amide II of BSA in the presence of SnSe nanoparticles. These shifts clearly indicate the involvement of –NH and disulfide linkages in the binding interactions of the protein with the nanoparticles.
O stretching vibrations of the peptide bond (belongs to amide I) and N–H stretching vibrations of amide A, respectively.34 The peak at 1630 cm−1 corresponds to short segment chains connecting α-helical segments of BSA.35 Also, a peak at ∼564 cm−1 was observed (in case of pure BSA), and could be attributed to the disulfide stretching vibrations.36 The strong resemblance of the FTIR spectra of pure BSA with the BSA–SnSe categorically indicates towards the significant coating of the former on the surface of the nanoparticles. Further, a red shift of ∼28 cm−1 and 46 cm−1 was observed in the disulphide peak (from 564 to 536 cm−1) and bending vibrations of amide III (from 1246 to 1200 cm−1), respectively in the FTIR spectrum of BSA–SnSe as compared to that of pure BSA. On the other hand, a blue shift of 27 cm−1 (from 3043 to 3070 cm−1) and 4 cm−1 (from 1520 to 1524 cm−1) was noticed in the N–H stretching vibrations of amide A and N–H bending vibration of amide II of BSA in the presence of SnSe nanoparticles. These shifts clearly indicate the involvement of –NH and disulfide linkages in the binding interactions of the protein with the nanoparticles.
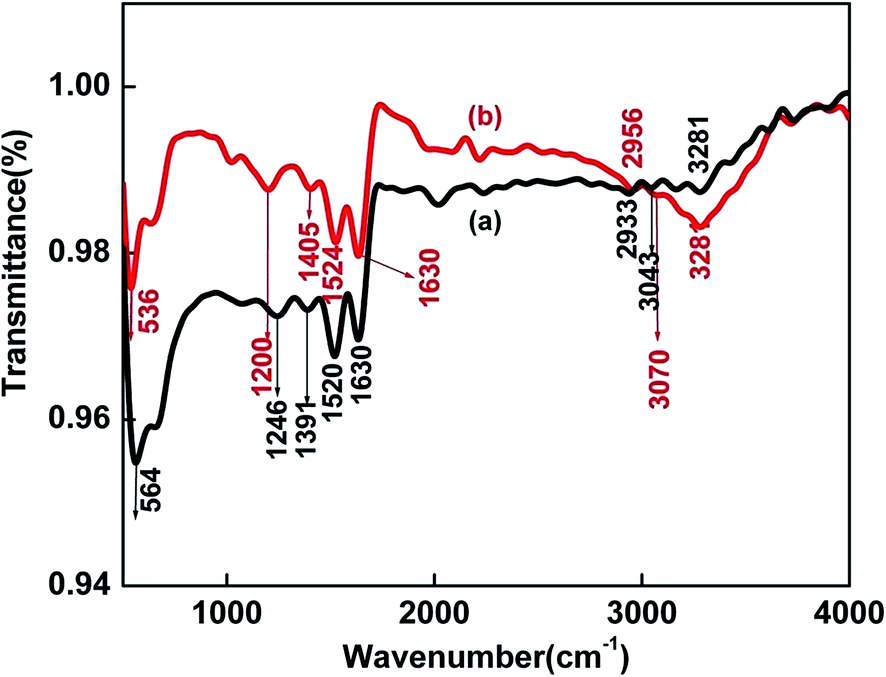 | ||
| Fig. 3 FTIR spectra of (a) pure BSA (b) BSA–SnSe. | ||
Apart from this, the conformational changes in the intrinsic structure of protein (in the presence of SnSe) are evident from the CD spectral analysis (discussed later).
To determine the influence of such structural perturbations on the morphology of as grown nanoparticles, TEM images were recorded. Fig. 4 represents the TEM images of SnSe nanoparticles at different time intervals.
 | ||
| Fig. 4 TEM images showing the (a) self-assembling of primary nanoparticles (image recorded within 5 minutes of the reaction); (b) formation of sheet like nanostructures (image recorded after ∼20 minutes of the reaction); (c) nanosheets of SnSe (bar dimensions = 200 nm) (image recorded after ∼2 hours of the reaction); (d) SAED pattern indicating the single crystalline nature of the nanosheets of SnSe. | ||
Interestingly, it can be observed that the primary nanoparticles formed at the initial stages of the reaction (Fig. 4a and b) were found to self-assemble into a combination of 2D square and rectangular sheet-like nanostructures (Fig. 4c). Some more TEM images showing the formation of nanosheets has been provided in the ESI (Fig. S3†). SEM measurements of the as grown nanomaterial were also carried out (shown in Fig. S4†), which also confirmed the formation of sheet like nanostructures. The average length and width of the sheets were found to be between 100 and 150 nm. Furthermore, the sheets were single crystalline in nature, as can be seen from the selected area electron diffraction (SAED) pattern shown in Fig. 4d. In general, layered crystal structures are obtained by using those stabilizing ligands which permit only lateral growth rather than vertical.15,37 The plausible role of protein structure in directing the as obtained self-assembly of SnSe nanoparticles into 2D structures might be explained as follows. It has been already mentioned in the experimental section that the precursor of Sn was first added to the protein solution. At this stage, the pH of the solution was determined to be ∼3.0. It is a well reported fact that BSA conformation is more open and expanded at lower pH values leading to the access of more disulfide bonds.38–40 Further, the impairment or break down of the hydrogen bonds eventually results into the transformation of α-helix into β-sheet structure of BSA.41 This was further substantiated by the CD spectral studies, wherein the negative band at ∼222 nm characterizing the α-helix structure of protein41 was shifted to ∼218 nm on adding the precursor solution of Sn (having pH ∼ 3.0) (see Fig. S2†). This peak at ∼218 nm signifies the formation of β-pleated sheet structure.41 The CD spectrum of the BSA solution having pH = 3.0 was similar to the solution containing BSA + Sn precursor solution. Subsequently, the addition of the Se precursor (Na2SeSO3) turned the color of the resulting solution to greenish yellow. At this stage, the primary SnSe nanoparticles formed undergo self-assembling, as was observed from Fig. 4a and b. Apparently, it can be anticipated that the β-sheet structure of BSA is most probably acting as a shape directing template for the self-orientation of primary nanoparticles into as obtained 2D sheet-like nanostructures.
Another important observation is the room temperature PL displayed by as grown SnSe nanosheets (shown in Fig. 5a), which has been reported by few researchers.20,42 The PL appeared to be the convolution of two bands i.e. Band-I at ∼425 nm and Band-II at ∼470 nm. These bands were found to be independent of excitation wavelengths and assigned to be originated from band-gap (BGPL) and trap/surface state emission (TSPL). The Band-I was attributed to the BGPL due to its close proximity from the excitonic peak position, while Band-II appeared to represent was TSPL due to its large stokes shift relative to the excitonic absorption peak. To further confirm their origins, time resolved PL decay studies (shown in Fig. 5b) were conducted at two emission wavelengths i.e. 420 and 470 nm.
 | ||
| Fig. 5 (a) Room temperature PL spectra; (b) time resolved PL decay curve (λexc = 374 nm) of SnSe nanosheets at two emission wavelengths (420 and 470 nm). | ||
The PL decay curves exhibited multi-exponential (tri-exponential) behavior, which were analyzed and fitted using eqn (2):
| I(t) = a1e−t/τ1 + a2e−t/τ2 + a3e−t/τ3 | (2) |
The average PL lifetime (〈τ〉) values were deduced using eqn (3):
 | (3) |
Such multi-exponential behavior of the binary semiconductor nanoparticles is widely known and has been reported by various researchers earlier.43–45 In general, most of these reports generally attribute such behavior to the diverse recombination paths owing to the varying degree of size, shape, surface defects/energy traps between the individual nanocrystals. Still, the origin and the explanation for this multi-exponential emission dynamics are not very clear and is currently a subject of debate. Nevertheless, the average lifetime (〈τ〉) (see Table 1) obtained by analyzing and fitting the decay traces at 420 and 470 nm were obtained as 2.1 and 4.0 ns, respectively. The corresponding χ2 values for the former and the later decay traces are 1.0 and 1.1, respectively. At large, the shorter life-time is accredited to the band gap excitonic recombination, while the longer lifetime is attributed to the involvement of surface states in the carrier recombination process.46 Therefore, these findings confirm our earlier assignments of Band-I and Band-II as BGPL and TSPL, respectively.
| λ em, nm | T 1, ns | B 1 (%) | T 2, ns | B 2 (%) | T 3, ns | B 3 (%) | χ 2 | 〈t〉, ns |
|---|---|---|---|---|---|---|---|---|
| 420 | 1.04 | 32 | 5.57 | 32 | 0.095 | 36 | 1.0 | 2.1 |
| 470 | 1.67 | 41 | 6.81 | 44 | 0.31 | 15 | 1.1 | ∼4.0 |
The room temperature PL of the SnSe nanosheets has scope for applications in bio-labeling and bio-imaging.47 Purposefully, the synthesized nanomaterial needs to be biocompatible. Recently, nanotoxicology has become an important field of research.48,49 Several groups have tried to reduce the level of cytotoxicity of fluorescent QDs like CdSe, which can be used as an appropriate alternative against organic dyes in bio-labeling.50 It has been shown that BSA coating render the QDs more biocompatible with reduced cytotoxicity rather than ZnS capping alone.50 In the present case, the cytotoxicity experiments carried out on CHO cells provided positive indication (see Fig. 6). There was no significant rise in toxicity compared to the control with the increase in the concentration of SnSe nanosheet colloidal solution. Up to a concentration of 100 μM, the viability remains almost constant at ∼90%. This opens up the possibility for the use of as synthesized nanomaterial in diagnostic and therapeutic purposes.
 | ||
| Fig. 6 Cytotoxic effects of increasing concentrations of the colloidal solution of SnSe nanosheets in CHO cells as determined by the MTT assay. Values are mean ± SEM of three experiments. | ||
The CV of the colloidal solution of SnSe nanosheets was carried out in TEABF4–acetonitrile mixture solution. The onset reduction potential appeared at −0.59 V, as can be envisaged from Fig. 7. To study the effect of Li ions, 0.1 (M) LiBF4 was added to the electrolyte. A large increase in reduction current was observed indicating that LiBF4 facilitates the electron transfer due to intercalation of Li+, as shown in the equation below.
| SnSe + 2Li+ + 2e = Li2Se + Sn | (4) |
 | ||
| Fig. 7 CV measurements using Ag/Ag+ as the reference electrode (a) acetonitrile, TEABF4 electrolyte mixture as blank (b) SnSe deposited on glassy carbon electrode in acetonitrile, TEABF4 electrolyte mixture (c) 0.1 (M) LiBF4 added to acetonitrile, TEABF4 mixture in the presence of SnSe. | ||
The reduction potential value was found to shift to −0.45 V in the presence of Li ions due to their intercalation within the SnSe lattice. At this stage, it is being mentioned here that a reduction potential of −0.97 and −0.91 V has been reported for SnSe nanosheets and nanoflowers, respectively, as prepared by using oleylamine and 1,10-phenanthroline at 260 °C.32 These values are higher than −1.06 and −1.4 V reported for SnSe nanocrystals.32,51
As compared to previously reported reduction potential values, our present value is higher thereby, indicating better thermodynamic feasibility for the reduction of SnSe. This demonstrates that the SnSe prepared using the present method gives better redox characteristics from the point of view of its application in Li-ion batteries. The material can be thought for use as an anode for Li-ion battery, after added modifications, which needs further investigation.
4. Conclusions
A purely green methodology was employed for the aqueous phase synthesis of SnSe nanosheets in the host matrix of BSA. Through time dependent HRTEM imaging, FTIR and circular dichroism studies, the mechanism behind the formation of SnSe nanosheets has been proposed. It was revealed that the initially formed SnSe nanoparticles self-assemble into single crystalline sheets under the influence of β-pleated sheet structure of BSA. Therefore, BSA is playing three roles simultaneously i.e. as a reducing agent, stabilizing agent and a shape directing template. The as grown SnSe nanomaterial was found to be photoluminescent and the PL spectra appeared to be the convolution of band-gap and trap/surface state emission. Cyclic voltammetry measurements indicate better thermodynamic feasibility for the reduction of SnSe and thereby, confer its potential application in lithium ion batteries. Cytotoxicity studies of as grown protein encapsulated SnSe nanomaterial revealed no toxic effects up to a concentration of 100 μM, which signifies the advantage of this methodology with a view point of their biomedical applications.Acknowledgements
The authors thank Dr A. K. Debnath, Dr N. Barooah, and Dr A. Kunwar for carrying out XPS, Circular dichroism and Cytotoxicity studies, respectively. Also, sincere thanks to Dr Shilpa Sawant for the cyclic voltammetry experiments and Dr A. D. Ballal and Shri S. Neogy for the TEM measurements. The authors thankfully acknowledge Mr Jitendra Nuwad and Dr S. Bhattacharya for the SEM measurements. The authors thank D. K. Palit, Head, RPCD and B. N. Jagatap, Director, Chemistry Group, BARC for their support and encouragement.References
- E. Hao, H. Sun, Z. Zhou, J. Liu, B. Yang and J. Shen, Chem. Mater., 1999, 11, 3096–3102 CrossRef.
- T. Trindade, P. O'Brien and N. L. Pickett, Chem. Mater., 2001, 13, 3843–3858 CrossRef.
- D. S. Bera, L. Qian, T. K. Tseng and P. H. Holloway, Materials, 2010, 3, 2260–2345 CrossRef.
- Y. Wang, J. Nanosci. Nanotechnol., 2008, 8, 1068–1091 Search PubMed.
- M. F. Frasco and N. Chaniotakis, Sensors, 2009, 9, 7266–7286 CrossRef PubMed.
- V. Lesnyak, N. Gaponik and A. Eychmuller, Chem. Soc. Rev., 2013, 42, 2905–2929 RSC.
- P. V. Kamat, J. Phys. Chem. C, 2008, 112, 18737–18753 Search PubMed.
- P. D. Antunez, J. J. Buckley and R. L. Brutchey, Nanoscale, 2011, 3, 2399–2411 RSC.
- M. A. Franzman, C. W. Schlenker, M. E. Thompson and R. L. Brutchey, J. Am. Chem. Soc., 2010, 132, 4060–4061 CrossRef PubMed.
- N. R. Mathews, Sol. Energy, 2012, 86, 1010–1016 CrossRef.
- K. M. Chung, D. Wamwangi, M. Woda, M. Wuttig and W. Bench, J. Appl. Phys., 2008, 103, 083523 CrossRef.
- S. Kanga, L. Jia, X. Li, Y. Yin, L. Li, Y. Guob and J. Mua, Colloids Surf., A, 2012, 406, 1–5 CrossRef.
- L. Zhao, S. Lo, Y. Zhang, H. Sun, G. Tan, C. Uher, C. Wolverton, V. P. Dravid and M. G. Kanatzidis, Nature, 2014, 508, 373–377 CrossRef PubMed.
- P. D. Antunez, J. J. Buckley and R. L. Brutchey, Nanoscale, 2011, 3, 2399–2411 RSC.
- D. D. Vaughn II, S. In and R. E. Schaak, ACS Nano, 2011, 11, 8852–8860 CrossRef PubMed.
- X. Ma, K. Cho and Y. Sung, CrystEngComm, 2014, 16, 5080–5086 RSC.
- L. Li, Z. Chen, Y. Hu, X. Wang, T. Zhang, W. Chen and Q. Wang, J. Am. Chem. Soc., 2013, 135, 1213–1216 CrossRef PubMed.
- W. J. Baumgardner, J. J. Choi, Y. Lim and T. Hanrath, J. Am. Chem. Soc., 2010, 132, 9519–9521 CrossRef PubMed.
- K. Jang, I. Lee, J. Xu, J. Choi, J. Jin, J. Park, H. Kim, G. Kim and S. Son, Cryst. Growth Des., 2012, 12, 3388–3391 Search PubMed.
- Z. Li, L. Peng, Y. Fang, Z. Chen, D. Pan and M. Wu, Radiat. Phys. Chem., 2011, 80, 1333–1336 CrossRef.
- H. S. Im, Y. Myung, Y. Cho, C. Kim, H. Kim, S. Back, C. Jung, D. M. Jang, Y. R. Lim, J. Park and J. Ahn, RSC Adv., 2013, 3, 10349–10354 RSC.
- F. K. Butt, M. Mirza, C. Cao, F. Idrees, M. Tahir, M. Safdar, Z. Ali, M. Tanveera and I. Aslam, CrystEngComm, 2014, 16, 3470–3473 RSC.
- M. Ahmed, A. Guleria, M. C. Rath, A. K. Singh, S. Adhikari and S. K. Sarkar, J. Nanosci. Nanotechnol., 2014, 14, 5730–5742 CrossRef PubMed.
- M. C. Rath, J. A. Mondal, D. K. Palit, T. Mukherjee and H. N. Ghosh, J. Nanomater., 2007, 2007, 1–7 CrossRef.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef PubMed.
- L. Ling, Q. Zhang, L. Zhu, C. F. Wang and S. Chen, RSC Adv., 2015, 5, 2155–2158 RSC.
- R. Indirajith, T. P. Srinivasan, K. Ramamurthi and R. Gopalakrishnan, Curr. Appl. Phys., 2010, 10, 1402–1406 CrossRef.
- K. J. John, B. Pradeep and E. Mathai, J. Mater. Sci., 1994, 29, 1581–1583 CrossRef.
- N. D. Boscher, C. J. Carmalt, R. G. Palgrave and I. D. Parkin, Thin Solid Films, 2008, 516, 4750–4757 CrossRef.
- K. G. S. Ranmohotti, H. Djieutedjeu and P. F. P. Poudeu, J. Am. Chem. Soc., 2012, 134, 14033–14042 CrossRef PubMed.
- H. Djieutedjeu, X. Zhou, H. Chi, N. Haldolaarachchige, K. G. S. Ranmohotti, C. Uher, D. Young and P. F. P. Poudeu, J. Mater. Chem. C, 2014, 2, 6199 RSC.
- L. Li, Z. Chen, Y. Hu, X. Wang, T. Zhang, W. Chen and Q. Wang, J. Am. Chem. Soc., 2013, 135, 1213–1216 CrossRef PubMed.
- W. Zhang, Z. Yang, J. Liu, L. Zhang, Z. Hui, W. Yu, Y. Qian, L. Chen and X. Liu, J. Cryst. Growth, 2007, 217, 157–160 CrossRef.
- K. Fu, K. Griebenow, L. Hsieh, A. M. Klibanov and R. Langer, J. Controlled Release, 1999, 58, 357–366 CrossRef PubMed.
- K. Murayama and M. Tomida, Biochemistry, 2004, 43, 11526–11532 CrossRef PubMed.
- Biopolymer Research Trends, ed. T. Nemeth, Nova Science Publishers, Inc., New York, 2008, pp. 22 and 29 Search PubMed.
- J. Son, J. H. Yu, S. Kwon, J. Lee, J. Joo and T. Hyeon, Adv. Mater., 2011, 23, 3214–3219 CrossRef PubMed.
- N. Kadi, N. Taulier, J. Y. Huérou, M. Gindre, W. Urbach, I. Nwigwe, P. C. Kahn and M. Waks, Biophys. J., 2006, 91, 3397–3404 CrossRef PubMed.
- P. J. Sadler and A. Tucker, Eur. J. Biochem., 1993, 212, 811–817 CrossRef PubMed.
- E. Katchalski, G. S. Benjaminan and V. Gross, J. Am. Chem. Soc., 1957, 79, 4096–4099 CrossRef.
- N. J. Greenfield, Nat. Protoc., 2007, 1, 2876–2890 CrossRef PubMed.
- F. K. Butt, C. Cao, W. S. Khan, Z. Ali, R. Ahmed, F. Idrees, I. Aslam, M. Tanveer, J. Li, S. Zaman and T. Mahmood, Adv. Mater. Phys. Chem., 2012, 137, 565–570 CrossRef.
- B. R. Fisher, H. J. Eisler, N. E. Stott and M. G. Bawendi, J. Phys. Chem. B, 2004, 108, 143 CrossRef.
- A. A. Salman, A. Tortschanoff, G. V. Zwan, F. V. Mourik and M. Chergui, Chem. Phys., 2009, 357, 96 CrossRef.
- A. F. V. Driel, I. S. Nikolaev, P. Vergeer, P. Lodahl, D. Vanmaekelbergh and W. L. Vos, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 75, 035329 CrossRef.
- J. Z. Zhang, J. Phys. Chem. B, 2000, 104, 7239 CrossRef CAS.
- W. J. Parak, T. Pellegrino and C. Plank, Nanotechnology, 2005, 16, R9–R25 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- M. Tarantola, D. Schneider, E. Sunnick, H. Adam, S. Pierrat, C. Rosman, V. Breus, C. Sonnichsen, T. Basche, J. Wegener and A. Janshoff, ACS Nano, 2009, 3, 213–222 CrossRef CAS PubMed.
- A. M. Derfus, W. C. W. Chan and S. N. Bhatia, Nano Lett., 2004, 4, 11–18 CrossRef CAS.
- S. Liu, X. Guo, M. Li, W. Zhang, X. Liu and C. Li, Angew. Chem., Int. Ed., 2011, 50, 12050–12053 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09448h |
| This journal is © The Royal Society of Chemistry 2015 |