
α-Chymotrypsin and l-acylase aided synthesis of 5-hydroxypipecolic acid via Jacobsen's hydrolytic kinetic resolution of epoxy amino acids†

Abstract
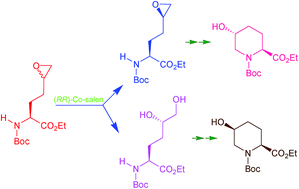
Diethyl malonate derivatives were used to synthesize racemic 2-amino-5-hexenoic acid. These racemic 2-amino-5-hexenoic acid (homoallylyglycine) derivatives were efficiently resolved aided by α-chymotrypsin or L-acylase, giving rise to L- and D-enantiomers. These isolated enantiomerically pure amino acids with tert-butoxycarbonyl (Boc) protection were oxidised with 3-chloroperbenzoic acid. The oxidation gave rise to inseparable diastereomeric epoxides due to a newly generated chiral center at the C5 carbon. The isolation of one of the diastereomeric epoxides was possible by selectively converting the remaining diastereomer into a dihydroxyl compound catalysed by Jacobsen's hydrolytic kinetic resolution (HKR). The isolated epoxide was regioselectively attacked by LiBr to give vicinal halohydrin, with bromide attacking the terminal C6 carbon. Boc deprotection of the halohydrin led to intramolecular cyclization by attack of free amine at the C6 carbon, generating a single isomer of 5-hydroxypipecolic acid which was effortlessly recovered in good yield after re-protection of the amine with the Boc group. Similarly the dihydroxyl compound isolated earlier was converted to a halohydrin with iodine at the C6 carbon. This was feasible by efficient regioselective mono-tosylation with catalytic Bu2SnO followed by iodine substitution. This was utilized to synthesize the 5-hydroxypipecolic acid derivative in the same described sequence consisting of Boc removal by acid treatment, cyclization, reprotection and purification. Finally the same sequence was repeated with the D-isomer and two diastereomers were isolated.
 Please wait while we load your content...
Please wait while we load your content...

