
CuFe2O4@PDA magnetic nanomaterials with a core–shell structure: synthesis and catalytic application in the degradation of methylene blue in water†
Su-dai Maab,
Jie Fengab,
Wen-jie Qinab,
Yu-yun Juab and
Xing-guo Chen*abc
aState Key Laboratory of Applied Organic Chemistry, Lanzhou University, Lanzhou, 730000, China. E-mail: chenxg@lzu.edu.cn; Fax: +86-931-8912582; Tel: +86-931-8912763
bDepartment of Chemistry, Lanzhou University, Lanzhou, 730000, China
cKey Laboratory of Nonferrous Metal Chemistry and Resources Utilization of Gansu Province, Lanzhou University, Lanzhou, 730000, China
First published on 10th June 2015
Abstract
In this paper, core–shell polydopamine (PDA)-encapsulated CuFe2O4 (CuFe2O4@PDA) magnetic nanoparticles (MNPs) were synthesized through in situ self-polymerization for the first time. The size of the core–shell product can be controlled by tuning the dopamine monomer concentration. The formation of a PDA layer effectively enhanced the catalytic performance and provided a large specific surface area which offered more active sites for the effective interaction. The as-synthesized CuFe2O4@PDA MNPs were characterized and their catalytic activity was evaluated using the degradation of methylene blue (MB) in the presence of H2O2 as a model reaction. The experimental results showed that MB could be degraded efficiently using CuFe2O4@PDA MNPs as a catalyst. Under the optimized conditions, the degradation efficiency of MB was above 97%. Furthermore, a possible reaction mechanism was discussed. Finally, the catalyst was used for effective degradation of MB in a Yellow River water sample, which indicates its potential for practical applications in water pollutant removal and environmental remediation.
1. Introduction
Rapid industrialization has led to an increased amount of discharged wastewater containing pollutants. Water pollution by organic dyes has become a serious environmental issue and has received remarkable attention. The undesirable and immoderate release of wastewater containing organic dyes has caused a pernicious impact, including destroying the balance of ecological systems, destruction of the food chain existing in water ecosystems and endangering animals and human beings,1–5 etc. MB is an important member of the thiazine class of dyes. It is most widely used in paper, textiles, plastics, food, leather and cosmetic to color products.6 However, acute exposure to MB will cause increased heart rate, shock, vomiting, Heinz body formation, jaundice, cyanosis, quadriplegia and tissue necrosis in humans.7–10 The development of a process for the efficient treatment of MB-polluted wastewater is therefore of utmost importance. The various conventional treatment methods, such as physical adsorption,11,12 chemical oxidation,13,14 biological degradation,15 photocatalytic degradation,16,17 and sonochemical technology18 have been developed for the removal of such contaminants in wastewater. However, they suffer from some disadvantages including incomplete removal, additional equipment and disposal problem of sludge, etc. So it is still of great significance to discover a relatively low cost, easy operation and higher activity materials for efficient removal of organic dyes.In recent years, the core–shell nanomaterials were developed as novel nanostructured catalyst to remove organic dyes. The core–shell structural particles showed some special properties such as large specific surface area, adjustable size and devisable chemical composition. Moreover, the core–shell structural particles which may combine the advantages of “core” and “shell” showed enhanced physical and chemical properties in optics, electronics, magnetics and catalysis.19–22 Thus, a lot of researches have been reported on the functional shells, including polymers, ligands, oxides or metals.23,24 Dopamine (DA), one of the important natural chemical neurotransmitters presented in various animals. It had the ability of adhering to almost all material surfaces to form thin and surface-adherent polydopamine (PDA) coating under mild condition.25–28 In addition, compared with other shell such as SiO2, Au, PDA shell displays the following characteristics: (1) the process of preparing a PDA coating seems to be simple, quick and ‘green’;29,30 (2) a robust interfacial binding force between the coating and the substrate through covalent bonds or other strong intermolecular interactions is easily adapted for a variety of materials without surface pretreatment; (3) PDA coating has plenty of functional groups such as amino and hydroxyl groups as well as π–π bonds, thus establishing further multiple modification.31–34 (4) The good electrochemical behavior of a PDA coating with π–π stacking interaction can accelerate the electron transfer. On the basis of excellent characteristics, PDA coating is an outstanding candidate to develop core–shell nanomaterials.
However, the application of some nanomaterials was restricted due to difficult separation. This reason made it challenging to recycle and reuse the nanomaterials efficiently, further affecting their decontamination efficiencies in dye removal. It is therefore crucial to look for stable, separated easily, and high catalytic activity nanomaterials from the viewpoint of practical application. The integration of PDA with superparamagnetic materials to form uniform core–shell nanocomposite particles has emerged as a viable solution. As an important kind of magnetic materials, copper ferrites (CuFe2O4) with good superparamagnetic nature as a magnetic core can make the catalysts efficiently separated from the reaction mixture with an external magnet, which prevents the loss of catalyst and renders the catalyst cost effective. Furthermore, CuFe2O4 nanoparticles possess excellent catalytic activity in various catalytic applications.35–39
As far as we know, the research on nanocompounds combining the advantages of the CuFe2O4 and PDA has not been reported. Hence, a facile and eco-friendly method for the fabrication of magnetically recyclable CuFe2O4@PDA MNPs with core–shell nanostructures was reported (Fig. 1). The size of the core–shell product was finely controlled by tuning the DA monomer concentration to form varying shell thickness of the PDA layer. Then, the CuFe2O4@PDA MNPs were employed to investigate its catalytic potential in the degradation of MB. Our results demonstrated that CuFe2O4@PDA MNPs possess excellent catalytic activity toward the degradation of MB, with advantages such as wide working pH range, environmentally friendly preparation, long-term stability and easily removed from the reaction system. It is expected to become a promising catalyst applied in water pollutant removal and environmental remediation.
 | ||
| Fig. 1 Schematic diagram of the synthetic strategy and application of CuFe2O4@PDA magnetic nanoparticles. | ||
2. Experimental
2.1. Materials
FeCl3·6H2O was purchased from Tianjin Shuangchuan Chemical Reagent Factory (Tianjin, China). Ethylene glycol and anhydrous ethanol were purchased from Tianjin Rionlon Bohua Pharmaceutical Chemical Co., Ltd. (Tianjin, China). Polyethylene glycol 20,000 was got from Xi'an Chemical Reagent Factory (Xi'an, China). Sodium acetate (NaAc), hydrochloric acid (HCl) and sodium hydroxide (NaOH) were got from Tianjin Guangfu Chemical Reagent Factory (Tianjin, China). 30% H2O2 (with extremely small amount of PO43−) and MB were purchased from Tianjin Chemical Reagent Factory (Tianjin, China). CuCl2·2H2O and tris(hydroxymethyl)aminomethane (tris) were obtained from Sinopharm Chemica Reagent Co. Ltd. Dopamine hydrochloride (DA) was obtained from Sigma Company. All chemicals were of analytical grade and used without any further purification. Ultrapure water was used throughout the whole experiment. The solution pH was adjusted using diluted HCl and NaOH solutions.2.2. Preparation of CuFe2O4 magnetic nanoparticles
CuFe2O4 magnetic nanoparticles (CuFe2O4 MNPs) were prepared according to the literature procedures.37 Briefly, CuCl2·2H2O (2.5 mmol) and FeCl3·6H2O (5 mmol) were dissolved in ethylene glycol (40 mL) to form a clear solution, followed by the addition of NaAc (3.6 g) and polyethylene glycol 20,000 (1.0 g). The mixture was stirred vigorously for 30 min at 60–70 °C and then sealed in a Teflon lined stainless steel autoclave (50 mL capacity). The autoclave was heated to and maintained at 200 °C for 8 h and allowed to cool to room temperature. The generated black CuFe2O4 MNPs were collected by magnetic separation, washed several times with ethanol and ultrapure water. The resulting solids were dried at 60 °C for 3 h under vacuum.2.3. Synthesis of CuFe2O4@PDA magnetic nanoparticles
To prepare CuFe2O4@PDA MNPs, 0.4 g of the as-prepared CuFe2O4 MNPs was dispersed in 200 mL of dopamine–tris solution (0.5 mg mL−1, pH = 8.5, 10 mM tris–HCl buffer), and allowed to proceed for 8 h under stirring at room temperature. The resultant product was separated and collected with a magnet, and then washed with ultrapure water three times and dried at 60 °C for 3 h under vacuum.2.4. Apparatus and characterization
A Tecnai G2F30 instrument was used to obtain the morphology and particle size of the CuFe2O4 MNPs and CuFe2O4@PDA MNPs. A Nicolet Nexus 670 Fourier transform infrared spectrometer (FT-IR) was used to demonstrate the chemical nature of the products in KBr pellets. An X-ray diffraction (XRD) pattern of CuFe2O4 MNPs were carried out with a X'pertpro Philips X-ray diffractometer with Cu Kα radiation (λ = 1.54056 Å) and scanning in the 2θ range of 10–80°. The magnetic property of the nanoparticles was measured at room temperature with a vibration sample magnetometer (VSM, Lakeshore cryotronics 730, USA). A TU-1901 double beam UV-Vis spectrophotometer (Beijing Purkine General Instrument Co. Ltd., China) was used to implement scan spectrum and measure the absorbance for analysis. Fluorescent measurements were carried out by a RF-5301PC fluorescence (FL) spectrophotometer with a 1 cm quartz cell (Shimadzu, Kyoto, Japan). Mass spectra were recorded on an Esquire 6000 Mass spectrometer (Bruker Daltonics, USA). Inorganic ions in solution were analyzed using an DIONEX ICS-1500 Ion Chromatography (DIONEX, USA). Zeta potential measurements of CuFe2O4@PDA MNPs were performed with a Malvern Nano-ZS apparatus.2.5. Evaluation of catalytic performance
The degradation of MB in the presence of H2O2 was chosen as a model reaction to evaluate the catalytic properties of the as-prepared CuFe2O4@PDA MNPs. During the degradation, the reactor of 50 mL capacity was immersed into a thermostated shaker. For typical experimental runs, 4 mg freshly prepared CuFe2O4@PDA MNPs was dispersed into 18 mL aqueous solution of MB (1.0 × 10−5 mol L−1). The degradation of MB was initiated by rapid adding 2 mL H2O2 (5 mol L−1) to the reaction solution. After the mixed solution was shaken for 30 min, a strong magnet was deposited at the bottom of the conical flask, and the CuFe2O4@PDA MNPs were isolated from the solution. The MB concentrations remained the aqueous solutions were measured at λmax = 656 nm. In addition, the adsorption experiment was carried out under the same conditions just without H2O2. After oscillating 30 min, the change in absorbance of the mixture was measured again. The adsorption efficiency and the degradation efficiency of MB in the CuFe2O4/CuFe2O4@PDA MNPs system were calculated according to the equations:| η1 = [(A0 − A1)/A0] × 100% |
| η2 = [(A1 − A2)/A1] × 100% |
For reusability, the catalysts were collected and washed with ultrapure water after the catalytic reaction finished and the above catalytic process was repeated for several times until obvious inactivation was acquired. The control experiments were carried out under the same conditions without catalyst and/or H2O2.
Besides, a 3 mg portion of CuFe2O4@PDA MNPs were dispersed in 6 mL of ultrapure water and stored for a month to test the stability of the PDA layer in aqueous solution. A 3 mg portion of CuFe2O4@PDA MNPs were dispersed in 6 mL of hydrochloric acid solution at pH 2 and pH 3 for 24 h to test the acid stability of the sample.
The Yellow River water was chosen as a practical environmental water sample for investigation. Before use, the sample was centrifuged (13![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm) and filtered through a 0.45 μm membrane to remove any suspended particles. Stock solution was prepared by dissolving MB in the water sample. The degradation experiment was carried out under the same conditions and same operation.
000 rpm) and filtered through a 0.45 μm membrane to remove any suspended particles. Stock solution was prepared by dissolving MB in the water sample. The degradation experiment was carried out under the same conditions and same operation.
3. Results and discussion
3.1. Characterization of CuFe2O4 and CuFe2O4@PDA MNPs
XRD analysis was used to identify the crystal structure of the CuFe2O4 MNPs. As shown in Fig. S1,† except some Cu impurity peaks, all peaks were indexed to be CuFe2O4 (JCPDS 77-0010). There were five obvious diffraction peak at 2θ values of 29.98°, 35.22°, 57.10°, 62.28° and 74.06°, corresponding to the (220), (311), (422), (511) and (440) crystal plane of the spinel structure with CuFe2O4 MNPs. The reason of existence of metallic copper was that the strong reducing capability of ethylene glycol used as solvent in the preparation of the CuFe2O4 MNPs.40The morphology and size of the as-prepared CuFe2O4 MNPs and CuFe2O4@PDA MNPs were determined by TEM. Fig. 2a clearly displayed that the CuFe2O4 MNPs were spherical and dispersive with an average size of about 150 ± 20 nm. As shown in Fig. 2b, a continuous layer, which exhibited a fine increment in brightness in comparison to the dark inner core, was clearly observed on the outer shell of the CuFe2O4 core. From Fig. 2a and b, it was very clear that the CuFe2O4@PDA MNPs were encapsulated with a typical core–shell structure. There was a clear interface between the PDA shell and the CuFe2O4 core, indicating a tight encapsulation. In this process, the size of the CuFe2O4@PDA particle can be easily controlled by tuning the DA monomer concentration. As shown in Fig. 3, when the size of the CuFe2O4 microspheres was fixed and the DA monomer concentration was changed from 0.5 mg mL−1 to 4 mg mL−1, the average thickness of the PDA shell was 15 nm, 24 nm, 32 nm, 55 nm and 95 nm, respectively. The typical EDX pattern of CuFe2O4@PDA MNPs in Fig. 3f illustrated the fractions of all the elements in the MNPs. The appearance of N element indicated the successful modification of PDA on CuFe2O4 MNPs.
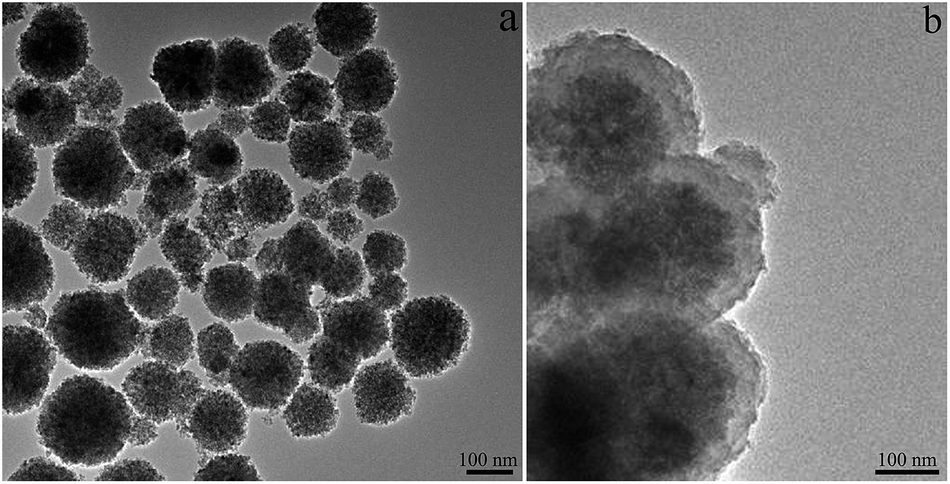 | ||
| Fig. 2 TEM images of CuFe2O4 MNPs (a) and CuFe2O4@PDA MNPs (b). | ||
 | ||
| Fig. 3 TEM images of the CuFe2O4@PDA MNPs based on CuFe2O4 sub-microspheres with different shell thicknesses: 15 nm (a), 24 nm (b), 32 nm (c), 55 nm (d) and 95 nm (e), corresponding to 0.5, 1, 2, 3, 4 mg mL−1 of DA monomer concentration, respectively. The EDX pattern of the CuFe2O4@PDA MNPs (f). | ||
FT-IR was employed to examine the surface composition of the synthesized the CuFe2O4 and CuFe2O4@PDA MNPs. As shown in Fig. 4a, the absorption band at 596 cm−1 and 417 cm−1 were ascribed to stretching vibration of tetrahedral complexes and octahedral complexes, respectively. The metal ions in ferrite were situated in two different sublattices owing to the geometrical configuration of the oxygen nearest neighbors.41 The adsorption peak at 3433 cm−1 represented the stretching mode of H2O molecules and OH groups. The absorption peak at 1626 cm−1 on spectrum referred to the vibration of remainder H2O in the sample.37 In Fig. 4b there were several new peaks compared with CuFe2O4 MNPs. The new peak at 1487 cm−1 belonged to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C stretching vibrations of aromatic ring in the PDA polymer. The absorption peak at 1576 cm−1 was attributed to N–H stretching and 1298 cm−1 was assigned to the C–O stretching of phenolic hydroxyl group.28,42 The peak at 3433 cm−1 of CuFe2O4@PDA MNPs was broader than that of CuFe2O4 MNPs, resulting from the overlapping of hydroxyls, water adsorbed in PDA polymer and amines of PDA. The FT-IR spectrum indicated that the PDA was indeed coated on the surface of the CuFe2O4 MNPs and the main structure of CuFe2O4 was not changed by the modification.
C stretching vibrations of aromatic ring in the PDA polymer. The absorption peak at 1576 cm−1 was attributed to N–H stretching and 1298 cm−1 was assigned to the C–O stretching of phenolic hydroxyl group.28,42 The peak at 3433 cm−1 of CuFe2O4@PDA MNPs was broader than that of CuFe2O4 MNPs, resulting from the overlapping of hydroxyls, water adsorbed in PDA polymer and amines of PDA. The FT-IR spectrum indicated that the PDA was indeed coated on the surface of the CuFe2O4 MNPs and the main structure of CuFe2O4 was not changed by the modification.
 | ||
| Fig. 4 FT-IR spectra of the CuFe2O4 MNPs (a) and CuFe2O4@PDA core–shell MNPs (b), respectively. | ||
Moreover, VSM measurement was employed to investigate the magnetic properties of the nanostructure. As shown in Fig. 5, a hysteresis loop of typical CuFe2O4 and CuFe2O4@PDA MNPs measured by sweeping the external field between 1.5 and 1.0 T at room temperature. In the VSM magnetization curves of the CuFe2O4 and CuFe2O4@PDA MNPs, there were no hysteresis, and the remanence and coercivity were negligible, indicating the superparamagnetism of these nanomaterials. Although the saturation magnetization value of CuFe2O4@PDA MNPs (39.15 emu g−1) was lower than CuFe2O4 MNPs (44.80 emu g−1) due to the existence of non-magnetic PDA coating, the CuFe2O4@PDA MNPs were readily separated from solution with a magnet due to their superparamagnetism and large saturation magnetization. All the above results revealed the successful synthesis of the CuFe2O4@PDA MNPs.
 | ||
| Fig. 5 Room-temperature magnetization hysteresis loops of the CuFe2O4 MNPs (a) and CuFe2O4@PDA MNPs (b). | ||
3.2. Evaluation of catalytic performance
In order to investigate the catalytic activity of the CuFe2O4@PDA MNPs, MB was used as a typical dye pollutant in the removal of organic pollutants for wastewater treatments. The degradation curve of the MB was shown in Fig. 6 by measuring the change of the absorbance at 665 nm. It showed that after the addition of the CuFe2O4@PDA MNPs, the characteristic absorbance of MB at 665 nm nearly disappeared, and the solution became colorless. It indicated that the CuFe2O4@PDA MNPs exhibited excellent catalytic activity in the degradation of MB. | ||
| Fig. 6 (a) UV-Vis absorption spectrum of MB degradation before and after addition of H2O2 and CuFe2O4@PDA MNPs. (b) Photograph of the degradation of MB by H2O2 in the presence of CuFe2O4@PDA MNPs. | ||
Moreover, the degradation efficiency of the CuFe2O4@PDA MNPs-H2O2 was 99% after 30 min (Fig. 7a), which was more effective than the CuFe2O4 MNPs-H2O2. The catalytic performance of CuFe2O4@PDA MNPs was about 668% and 434% higher than that of CuFe2O4 MNPs at 2 min and 5 min, respectively (Fig. S2†). However, no obvious decolorization was observed under the conditions of H2O2 only. The above results indicated that the core of the CuFe2O4 MNPs itself owned catalytic property. But the PDA layer can dramatically enhance the catalytic performance in MB degradation. The enhanced catalytic activity of CuFe2O4@PDA MNPs was probably caused by the synergistic effect between CuFe2O4 MNPs and PDA layer. In this process, MB was more easily absorbed on the surface of the CuFe2O4@PDA MNPs than CuFe2O4 MNPs.
 | ||
| Fig. 7 (a) Time-dependent degradation efficiency of MB in systems of MB + H2O2, CuFe2O4@PDA + MB + H2O2 and CuFe2O4 + MB + H2O2. (b) Time-dependent adsorption efficiency of MB in systems of CuFe2O4 + MB and CuFe2O4@PDA + MB. | ||
In order to further verify the adsorption effect of PDA layer, the adsorptions experiment of CuFe2O4@PDA MNPs and CuFe2O4 MNPs were also studied (Fig. 7b). There were nearly no decolorization in the presence of CuFe2O4 MNPs after 30 min, which demonstrated the CuFe2O4 MNPs could not adsorb MB. However, in the CuFe2O4@PDA-MB system, MB could be removed about 40% in the same time. Consequently, in these two parallel tests, the significant discrepancy was mainly caused by adsorption capacity of PDA layer. It is noted that there were three aspects may contribute to the adsorption of PDA layers and MB: (a) because of the large amount of functional groups (amino and catechol groups) on the surface of PDA layer, π–π interaction and hydrogen bonding induced the adsorption of MB toward the PDA surface.43–45 (b) The PDA layer also provided a large specific surface area and active sites for this process. (c) The electrostatic interactions favoured the efficient enrichment of the MB molecules on the PDA surfaces. To demonstrate this effect, the zeta potential of CuFe2O4@PDA MNPs was determined to be negatively charged in a wide pH range (Fig. S3†), suggesting strong electrostatic interactions with positively charged MB (Fig. S4†). To further confirm the presence of electrostatic interactions, the influence of pH on the adsorption performance of CuFe2O4@PDA MNPs was investigated (Fig. S5†). It was evident that MB was absorbed by the CuFe2O4@PDA MNPs faster at higher pH, which was most likely attributed to the stronger electrostatic interactions under higher pH. As confirmed by zeta potential measurement of CuFe2O4@PDA MNPs, the MNPs showed more negatively charged at higher pH. All of the evidences suggested that the PDA layer showed efficient adsorption of MB in addition to the high catalytic activity. As a result, the local concentration of MB at PDA surfaces was much higher than that in bulk solution. Therefore, the rate of the catalytic reaction and the performance of the catalysts were significantly improved.
3.3. Effect of operating parameters on MB degradation
 | ||
| Fig. 8 Effects of reaction conditions on MB degradation efficiency. (a) Dopamine monomer concentration. (b) The CuFe2O4@PDA MNPs amount. (c) H2O2 concentration. (d) pH. | ||
In summary, the optimized conditions for MB degradation using the CuFe2O4@PDA-H2O2 system were as follows: 4 mg CuFe2O4@PDA MNPs, 0.5 mol L−1 H2O2, pH 6.0, temperature 30 °C, oscillation time 30 min. Under optimal conditions, the MB could be removed completely, and the degradation efficiency was above 97%.
3.4. The reusability and the stability of CuFe2O4@PDA MNPs
The reusability and stability of catalyst are crucial properties to evaluate the catalytic performance. Therefore, the reusability of the as-prepared CuFe2O4@PDA MNPs was explored by checking the cycle number dependence of MB degradation loading the same CuFe2O4@PDA MNPs. Then the CuFe2O4@PDA MNPs were carefully collected after each cycle and reversibly reused in the identical catalytic system. As shown in Fig. S7,† the catalyst could be successfully recycled and reused for five successive cycles with a degradation efficiency of >85%.Furthermore, as shown in Fig. S8a,† after a month of soaking in aqueous solution, the PDA layers were still intact coated on the CuFe2O4 cores and the morphology remained unchanged. Even dispersed under a strong acid environment (pH 2 and pH 3) for over 24 h, the TEM images (Fig. S8b and c†) showed that the CuFe2O4@PDA MNPs still maintained the distinct core–shell structure. It is indicated that the robust PDA layer could effectively protect the CuFe2O4 core under strong acid conditions and show good stability in aqueous environment over a reasonably long period of time. Therefore, the magnetically separable CuFe2O4@PDA MNPs have outstanding recyclable and stable performance. It is expected to be used for a promising efficient catalyst in wastewater treatment.
3.5. Kinetics of the degradation of MB
Fig. 9 showed the time-dependent UV-Vis spectra change of MB catalyzed with CuFe2O4@PDA MNPs. The absorbance peak at 665 nm gradually attenuated during the degradation process and finally disappeared. Furthermore, the degradation rate was evaluated. For the MB degradation reaction, the ratio of the concentration ct of MB at time t to its initial value c0 at t = 0 were directly given by the ratio of the respective absorbance At/A0 (A represents the absorbance at 665 nm). Because H2O2 was in excess (cMB/cH2O2 = 9/500000) and its concentration was considered as a constant during the reaction process, the reaction kinetics could be treated as pseudo-first-order:| dct/dt = −kappct |
| ln(ct/c0) = ln(At/A0) = −kappt |
 | ||
| Fig. 9 The time-dependent UV-Vis absorption spectra change for the degradation process of MB. The illustration shows the relationship between ln(At/A0) and reaction time (t) for the MB reduction. | ||
Although the previous reported catalysts loading of gold or silver nanoparticles could also exhibit great catalytic activity for the degradation of MB,43,47 using expensive and scarce noble metals limited the widespread application of these catalysts.48 By contrast, the CuFe2O4@PDA MNPs were low-cost and affordable. Furthermore, the CuFe2O4@PDA MNPs exhibited other advantages, such as the wider working pH range, long-term stability, operational stability and higher catalytic efficiency compared with the analogous catalysts.18,49,50
3.6. Possible reaction mechanism
According to the above discussions, the degradation mechanism of MB was investigated and proposed. Obvious decolorization of MB was observed under the conditions of CuFe2O4@PDA MNPs and H2O2. In order to validate the reason of decolorization, the UV-Vis spectra were first inspected. Fig. 10 showed that after the addition of the CuFe2O4@PDA MNPs, the adsorption peak of 665 nm and 291 nm, which respectively represented the characteristic adsorption of conjugated structure and substituent benzene of MB, both vanished; a new strong adsorption peak (λ = 251 nm), which represented the characteristic adsorption of aromatic structure of catalytic degradation product, appeared.43,44 It indicated that the destruction of the whole MB molecular or the chromophore of MB. In order to further determine intermediates and final products, solutions of the MB and the MB was catalyzed by CuFe2O4@PDA MNPs were analyzed using ESI-MS (Fig. S9 and S10†). It is noteworthy that new fragments of m/z 228, 191, 131 were detected, demonstrating 3,7-diamino-phenothiazine-5-ium, 2,5-diaminobenzenesulfonic acid and DL-norleucine were formed in degradation process (Table S1†). The generation pathways of these fresh intermediates were speculated,51–53 as shown in Fig. S11.† Most Cl− may be ionized during the dissolution of MB and existed in the detached state. N–CH3 with the lowest bond energy was first broken. Then C–S and C–N were broken, C–S transformed into C–SO3H. C–NH2 bond in the remaining structure was broken and C–SO3H transformed into C–OH in the following degradation. The DL-norleucine generated as the ring in 1-amino-3,4-dihydroxybenzene opened. Mass spectra data provided overwhelming evidence of the MB degradation, which was in accordance with the results of UV-Vis spectra. Additionally, the ion chromatography was used to further determine ions in resultant degradation solution. As shown in Fig. S12,† Cl−, NO2−, NO3−, SO42− were detected. The generation of NO2−, NO3− and SO42− further manifested C–S and C–N bond gradually cleavage in the degradation process.52,53 These overall results confirmed that decolorization of MB was due to the MB was degraded by CuFe2O4@PDA MNPs as a catalyst in the presence of H2O2. | ||
| Fig. 10 Successive UV-Vis spectra of MB solution (black curve) and resultant solution after degraded by CuFe2O4@PDA MNPs (red curve). | ||
Based on the intermediate and final products detected, the degradation mechanism for MB is analyzed and described, as shown in Fig. S13.† The MB degradation in CuFe2O4@PDA-H2O2 system was mainly due to the synergistic effect of the CuFe2O4 core catalysis and PDA shell adsorption capacity. When the CuFe2O4@PDA MNPs were added and used for catalysis, H2O2 molecules and MB were adsorbed on the surface of CuFe2O4@PDA MNPs. The surface-adsorbed H2O2 molecules were activated to generate the ˙OH, which further react with adsorbed MB to initiate the degradation and/or diffuse into the solution to attack MB molecules near the CuFe2O4@PDA/solution interface. Furthermore, the PDA layer with π–π stacking interaction and the CuFe2O4 core with ‘dn’ (n = 5–9) electronic configuration could accelerate the electron transfer.37,54 The faster electron transferred on catalyst surface, the faster the reaction processed. To evidence this assumption, a fluorescence technique was used to detect of the produced ˙OH radicals by adding the fluorescent probe terephthalic acid into the CuFe2O4@PDA-H2O2 system, where terephthalic acid could easily react with ˙OH to yield a strongly fluorescent product 2-hydroxy terephthalic acid.55,56 It was clearly shown in Fig. S14† that fluorescence intensity enhanced gradually with the increasing concentration of the CuFe2O4@PDA MNPs, which suggested that the amount of generated ˙OH increased as CuFe2O4@PDA MNPs increased. However, no fluorescence intensity was observed in the absence of H2O2. All of the above results indicated that H2O2 molecules were adsorbed on the surface of the CuFe2O4@PDA MNPs and then activated by the CuFe2O4@PDA MNPs to generate reactive oxygen species. All the evidences suggested that the synergistic mechanism was feasible.
3.7. The application
Finally, in order to verify whether the nanocomposite could be applied to environmental water, the Yellow River water in Lanzhou section was collected and used as a practical sample. The Yellow River water formed a blue solution after being spiked with MB as a pollutant. Then 4 mg of the CuFe2O4@PDA MNPs was added to this water in the presence of H2O2. Satisfactory results were obtained that the solution became colorless, and the degradation efficiency was still above 97%. The CuFe2O4@PDA MNPs were then easily separated by a magnet and could be reused for further reactions. Therefore, the CuFe2O4@PDA MNPs were successfully used as a catalyst for the degradation of MB for complex environmental water samples and the proposed method was reliable.4. Conclusions
In summary, a simple method to prepare polydopamine modified CuFe2O4 MNPs was proposed. The PDA was directly grafted onto the CuFe2O4 using one step self-polymerization reaction to form well-defined core–shell nanostructures. The CuFe2O4@PDA MNPs exhibited excellent catalytic activity for the degradation of MB in the presence of H2O2. The CuFe2O4@PDA MNPs could be easily separated by a magnet after the catalytic reaction. In addition, the significant synergistic effect of the CuFe2O4 core catalysis and PDA shell adsorption capacity was observed. MB and H2O2 could be absorbed on the surface of the CuFe2O4@PDA MNPs. The surface-adsorbed H2O2 molecules were activated to generate the ˙OH, then it further reacted with adsorbed MB to initiate the degradation. The formation of PDA layer effectively enhanced the catalytic performance and protected the CuFe2O4 core to improve the stability. The versatile PDA polymer coating on the CuFe2O4 core also allowed the further surface functionalization for the development of multifunctional nanomaterials. Because of its merits such as wide working pH range, simple preparation, long-term stability, good recyclability and high catalytic performance, the CuFe2O4@PDA MNPs as potential catalysts will be facilitated to apply in various fields such as environmental protection, bioseparator, biosensor, and so on.Acknowledgements
The authors are grateful for financial support from the National Natural Science Foundation of China (no. 21375053) and Special Doctorial Program Fund from the Ministry of Education of China (no. 20130211110039).References
- T. Warang, N. Patel, R. Fernandes and N. Bazzanella, Appl. Catal., B, 2013, 132, 204–211 CrossRef PubMed.
- V. Vimonses, B. Jin and C. W. Chow, J. Hazard. Mater., 2010, 177, 420–427 CrossRef CAS PubMed.
- M. Doğan, H. Abak and M. Alkan, J. Hazard. Mater., 2009, 164, 172–181 CrossRef PubMed.
- B. S. Kaith, J. Dhiman and J. K. Bhatia, RSC Adv., 2015, 5, 39771–39784 RSC.
- Y. Y. Lau, Y. S. Wong, T. T. Teng, N. Morad, M. Rafatullah and S. A. Ong, RSC Adv., 2015, 5, 34206–34215 RSC.
- L. J. Yang, Y. Y. Zhang, X. Y. Liu, X. Q. Jiang, Z. Z. Zhang, T. T. Zhang and L. Zhang, Chem. Eng. J., 2014, 246, 88–96 CrossRef CAS PubMed.
- M. T. Uddin, M. A. Islam, S. Mahmud and M. Rukanuzzaman, J. Hazard. Mater., 2009, 164, 53–60 CrossRef CAS PubMed.
- M. Rafatullah, O. Sulaiman, R. Hashim and A. Ahmad, J. Hazard. Mater., 2010, 177, 70–80 CrossRef CAS PubMed.
- L. Li, X. L. Liu, M. Gao, W. Hong, G. Z. Liu, L. Fan, B. Hu, Q. H. Xia, L. Liu, G. W. Song and Z. S. Xu, J. Mater. Chem. A, 2014, 2, 1795–1801 CAS.
- H. T. Wang, H. Y. Ma, W. Zheng, D. D. An and C. Z. Na, ACS Appl. Mater. Interfaces, 2014, 6, 9426–9434 CAS.
- V. J. Vilar, C. Botelho and R. A. Boaventura, J. Hazard. Mater., 2007, 147, 120–132 CrossRef CAS PubMed.
- D. Özer, G. Dursun and A. Özer, J. Hazard. Mater., 2007, 144, 171–179 CrossRef PubMed.
- L. Yue, K. H. Wang, J. B. Guo, J. L. Yang, X. Luo, J. Lian and L. Wang, J. Ind. Eng. Chem., 2014, 20, 725–731 CrossRef CAS PubMed.
- R. Prihod'ko, I. Stolyarova, G. Gündüz, O. Taran, S. Yashnik, V. Parmon and V. Goncharuk, Appl. Catal., B, 2011, 104, 201–210 CrossRef PubMed.
- O. Olukanni, A. Osuntoki, D. Kalyani, G. Gbenle and S. Govindwar, J. Hazard. Mater., 2010, 184, 290–298 CrossRef CAS PubMed.
- M. Wu, J. Liu, J. Jin, C. Wang, S. Z. Huang, Z. Deng, Y. Li and B. L. Su, Appl. Catal., B, 2014, 150, 411–420 CrossRef PubMed.
- Y. C. Hsiao, T. F. Wu, Y. S. Wang, C. C. Hu and C. Huang, Appl. Catal., B, 2014, 148, 250–257 CrossRef PubMed.
- N. Wang, L. H. Zhu, D. L. Wang, M. Q. Wang, Z. F. Lin and H. Q. Tang, Ultrason. Sonochem., 2010, 17, 526–533 CrossRef CAS PubMed.
- F. Caruso, X. Shi, R. Caruso and A. Susha, Adv. Mater., 2001, 13, 740–744 CrossRef CAS.
- S. M. Marinakos, J. P. Novak, L. C. Brousseau, A. B. House, E. M. Edeki, J. C. Feldhaus and D. L. Feldheim, J. Am. Chem. Soc., 1999, 121, 8518–8522 CrossRef CAS.
- Y. Lu, Y. D. Yin, B. T. Mayers and Y. Xia, Nano Lett., 2002, 2, 183–186 CrossRef CAS.
- P. Tartaj and C. J. Serna, J. Am. Chem. Soc., 2003, 125, 15754–15755 CrossRef CAS PubMed.
- L. Y. Hao, C. L. Zhu, W. Q. Jiang, C. N. Chen, Y. Hu and Z. Y. Chen, J. Mater. Chem., 2004, 14, 2929–2934 RSC.
- Z. M. Liu, Y. L. Liu, H. F. Yang, Y. Yang, G. L. Shen and R. Q. Yu, Anal. Chim. Acta, 2005, 533, 3–9 CrossRef CAS PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- Q. Ye, F. Zhou and W. M. Liu, Chem. Soc. Rev., 2011, 40, 4244–4258 RSC.
- B. Fei, B. T. Qian, Z. Y. Yang, R. H. Wang, W. C. Liu, C. L. Mak and J. H. Xin, Carbon, 2008, 46, 1795–1797 CrossRef CAS PubMed.
- T. Zeng, X. L. Zhang, H. Y. Niu, Y. R. Ma, W. H. Li and Y. Q. Cai, Appl. Catal., B, 2013, 134, 26–33 CrossRef PubMed.
- H. Lee, Y. Lee, A. R. Statz, J. Rho, T. G. Park and P. B. Messersmith, Adv. Mater., 2008, 20, 1619–1623 CrossRef CAS PubMed.
- Y. Y. Li, C. Qin, C. Chen, Y. C. Fu, M. Ma and Q. J. Xie, Sens. Actuators, B, 2012, 168, 46–53 CrossRef CAS PubMed.
- W. Zhang, Y. Tang, J. Liu, Y. J. Ma, L. Jiang, W. Huang, F. W. Huo and D. B. Tian, J. Mater. Chem. B, 2014, 2, 8490–8495 RSC.
- Y. F. Wei, J. H. Kong, L. P. Yang, L. Ke, H. R. Tan, H. Liu, Y. Z. Huang, X. W. Sun, X. H. Lu and H. J. Du, J. Mater. Chem. A, 2013, 1, 5045–5052 CAS.
- M. Martin, P. Salazar, R. Villalonga, S. Campuzano, J. M. Pingarŕon and J. L. Gonźalez-Mora, J. Mater. Chem. B, 2014, 2, 739–746 RSC.
- N. Y. Wang, Y. Liu, Z. W. Qiao, L. Diestel, J. Zhou, A. Huang and J. Caro, J. Mater. Chem. A, 2015, 3, 4722–4728 CAS.
- Y. B. Wang, H. Y. Zhao, M. F. Li, J. Q. Fan and G. H. Zhao, Appl. Catal., B, 2014, 147, 534–545 CrossRef CAS PubMed.
- Z. H. Chonco, L. Lodya, M. Claeys and E. van Steen, J. Catal., 2013, 308, 363–373 CrossRef CAS PubMed.
- J. Feng, L. Su, Y. H. Ma, C. L. Ren, Q. Guo and X. G. Chen, Chem. Eng. J., 2013, 221, 16–24 CrossRef CAS PubMed.
- D. Kundu, N. Mukherjee and B. C. Ranu, RSC Adv., 2013, 3, 117–125 RSC.
- S. M. Baghbanian and M. Farhang, RSC Adv., 2014, 4, 11624–11633 RSC.
- H. Deng, H. Y. Chen and H. Li, Mater. Chem. Phys., 2007, 101, 509–513 CrossRef CAS PubMed.
- R. K. Selvan, C. Augustin, L. J. Berchmans and R. Saraswathi, Mater. Res. Bull., 2003, 38, 41–54 CrossRef CAS.
- P. An, F. Zuo, Y. P. Wu, J. H. Zhang, Z. H. Zheng, X. B. Ding and Y. X. Peng, Chin. Chem. Lett., 2012, 23, 1099–1102 CrossRef CAS PubMed.
- Y. J. Xie, B. Yan, H. L. Xu, J. Chen, Q. X. Liu, Y. H. Deng and H. B. Zeng, ACS Appl. Mater. Interfaces, 2014, 6, 8845–8852 CAS.
- A. J. Ma, Y. J. Xie, J. Xu, H. B. Zeng and H. L. Xu, Chem. Commun., 2015, 51, 1469–1471 RSC.
- H. L. Zhang, X. C. Li, G. H. He, J. J. Zhan and D. Liu, Ind. Eng. Chem. Res., 2013, 52, 16902–16910 CrossRef CAS.
- J. Z. Jiang, J. Zou, L. H. Zhu, L. Huang, H. P. Jiang and Y. X. Zhang, J. Nanosci. Nanotechnol., 2011, 11, 4793–4799 CrossRef CAS PubMed.
- J. Hu, Y. L. Dong, Z. U. Rahman, Y. H. Ma, C. L. Ren and X. G. Chen, Chem. Eng. J., 2014, 221, 16–24 Search PubMed.
- H. T. Wang, Z. X. Dong and C. Z. Na, ACS Sustainable Chem. Eng., 2013, 1, 746–752 CAS.
- H. Wang and Y. M. Huang, J. Hazard. Mater., 2011, 191, 163–169 CrossRef CAS PubMed.
- Z. Zhang, J. H. Hao, W. S. Yang, B. P. Lu, X. Ke, B. L. Zhang and J. L. Tang, ACS Appl. Mater. Interfaces, 2013, 5, 3809–3815 CAS.
- A. Chithambararaj, N. S. Sanjini, A. C. Bose and S. Velmathi, Catal. Sci. Technol., 2013, 3, 1405–1414 CAS.
- Q. Wang, S. L. Tian and P. Ning, Ind. Eng. Chem. Res., 2014, 53, 643–649 CrossRef CAS.
- Q. Wang, S. L. Tian, J. Long and P. Ning, Catal. Today, 2014, 224, 41–48 CrossRef CAS PubMed.
- Y. X. Wang, S. H. Wang, H. Y. Niu, Y. R. Ma, T. Zeng, Y. Q. Cai and Z. Meng, J. Chromatogr. A, 2013, 1283, 20–26 CrossRef CAS PubMed.
- T. Hirakawa and Y. Nosaka, Langmuir, 2002, 18, 3247–3254 CrossRef CAS.
- L. Su, J. Feng, X. M. Zhou, C. L. Ren, H. H. Li and X. G. Chen, Anal. Chem., 2012, 84, 5753–5758 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra09114d |
| This journal is © The Royal Society of Chemistry 2015 |