
Bi-modal cancer treatment utilizing therapeutic ultrasound and an engineered therapeutic nanobubble
Abstract
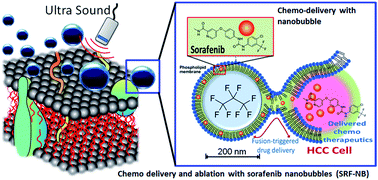
Herein we report a bi-modal cancer treatment technique using therapeutic ultrasound in the presence of a novel sorafenib loaded nanobubble for hepatocellular carcinoma. Therapeutic ultrasound was used for improving fusion-triggered drug release from nanobubbles leading to enhanced cytotoxicity in HepG2 cells by greater than two folds improvement in IC50 values. This strategy could also be made compatible with catheter-based devices to derive a highly localized treatment approach, a concept that might be extendable to metastatic cancers as well.
 Please wait while we load your content...
Please wait while we load your content...

