
Synthesis and characterization of glucosulfonic acid supported on Fe3O4 nanoparticles as a novel and magnetically recoverable nanocatalyst and its application in the synthesis of polyhydroquinoline and 2,3-dihydroquinazolin-4(1H)-one derivatives†
Abstract
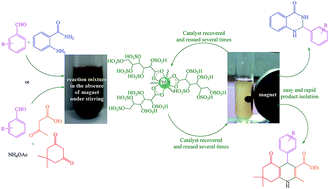
Glucosulfonic acid immobilized on Fe3O4 magnetic nanoparticles (GSA@MNPs) have been reported as an efficient and magnetically reusable nanocatalyst for the clean and one-pot synthesis of polyhydroquinoline and 2,3-dihydroquinazolin-4(1H)-one derivatives, with short reaction times in good to high yields in ethanol. After completing the reactions the catalyst was easily separated from the reaction mixture using an external magnetic field and reused for several consecutive runs without significant loss of catalytic efficiency. This new catalyst was characterized using FT-IR spectroscopy, TGA, XRD, VSM, TEM, EDS and SEM.
 Please wait while we load your content...
Please wait while we load your content...

