
Electroless deposition of Ni3P–Ni arrays on 3-D nickel foam as a high performance anode for lithium-ion batteries
Abstract
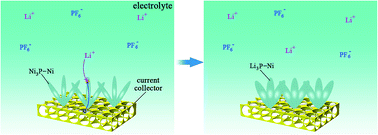
We successfully prepared advanced Ni3P–Ni array electrodes for Li-ion batteries (LIBs) by electroless deposition on 3-D nickel foam. The array structure of Ni3P–Ni can accommodate volume changes during the lithiation/de-lithiation process and promote high-rate capability because the interspaces in such structure can act as ideal volume expansion buffers. It shows excellent electrochemical performance as anode material for LIBs.
 Please wait while we load your content...
Please wait while we load your content...

