
Poly(dimethylsiloxane) (PDMS) surface patterning by biocompatible photo-crosslinking block copolymers†
Keita Kurodaa,
Hiromi Miyoshib,
Shota Fujiic,
Tomoyasu Hiraic,
Atsushi Takaharacd,
Aiko Nakaoe,
Yasuhiko Iwasakif,
Kenichi Morigakig,
Kazuhiko Ishiharah and
Shin-ichi Yusa*a
aDepartment of Materials Science and Chemistry, University of Hyogo, 2167 Shosha, Himeji, Hyogo 671-2280, Japan. E-mail: yusa@eng.u-hyogo.ac.jp; Fax: +81-79-267-4954; Tel: +81-79-266-8868
bRIKEN Center for Advanced Photonics, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
cGraduate School of Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
dInstitute for Materials Chemistry and Engineering, Kyushu University, 744 Motooka, Nishi-ku, Fukuoka 819-0395, Japan
eNishina Center for Accelerator-Based Science, Nuclear Spectroscopy Laboratory, RIKEN, 2-1 Hirosawa, Wako, Saitama 351-0198, Japan
fFaculty of Chemistry, Materials and Bioengineering, Kansai University, 3-3-35 Yamate, Suita, Osaka 564-8660, Japan
gResearch Center for Environmental Genomics, Kobe University, 1-1 Rokkodai, Nada, Kobe 657-8501, Japan
hDepartment of Materials Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo 113-8656, Japan
First published on 20th May 2015
Abstract
Poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) possesses protein antifouling properties. Diblock copolymers (PMPC120-b-P(TSM/CEAx)y) composed of a PMPC block and random copolymer block with 3-(tris(trimethylsiloxy)silyl)propyl methacrylate (TSM) and 2-cinnamoylethyl acrylate (CEA) were prepared via reversible addition–fragmentation chain transfer (RAFT) radical polymerization. A thin film of PMPC120-P(TSM/CEAx)y formed on the surface of the poly(dimethylsiloxane) (PDMS) substrate due to physical adsorption of the TSM units to poly(dimethylsiloxane) (PDMS) and photo-crosslinking of the CEA units. A lattice pattern of PMPC120-P(TSM/CEAx)y on the PDMS surface was prepared using UV irradiation through a photomask. PMPC120-P(TSM/CEAx)y-coated PDMS demonstrated protein antifouling activity. Cell patterning could be achieved by culturing on the PMPC-patterned PDMS substrate.
1. Introduction
Poly(dimethylsiloxane) (PDMS) is used for biomedical and biological applications such as catheters, contact lenses, micro-flow channels, and cell culture substrates because of its low toxicity, flexibility, processability, mechanical properties, and gas permeability.1–4 However, when used for biomedical and bioanalytical devices, PDMS can absorb proteins, which can adversely affect the function of the device.PDMS surface modifications to suppress protein adhesion also are necessary for cell patterning techniques, such as tissue engineering, regeneration, and maintenance. The cell culture location, which controls the adhesion and migration of cells, can be determined through the design of the culture substrate surface. In addition, the culture location affects cell proliferation, differentiation, and molecular signaling.5 Attempts have been made to clarify cell function and adhesion behavior by cell patterning.6–11 It is possible that cell patterning can be achieved through cell cultures in specific locations on the substrate.12,13
The pendant phosphorylcholine groups in poly(2-methacryloyloxyethyl phosphorylcholine) (PMPC) possess the same chemical structure as the hydrophilic part of phosphatidylcholine that forms a cell membrane.14,15 Hydrophilic PMPC possesses excellent biocompatibility and anti-thrombosis activity. A PMPC coating on the surface of various materials led to new functions, such as protein antifouling, high wettability, and low friction. In fact, PMPC coatings have been used in medicine for devices such as artificial hip joints to prevent wear over the long term.16–19 2-Methacryloyloxyethyl phosphorylcholine (MPC) can be radical-copolymerized with various comonomers to add functionalities to the obtained MPC copolymer. Although poly(ethylene glycol) (PEG) is a very useful to suppress protein adhesion, its applications are limited in some circumstances by its characteristics. Sometimes PEG can autoxidize in the presence of oxygen and transition metal ions. Particularly, the terminal hydroxyl group in PEG can be oxidized to aldehydes.20
Recently, various methods have been reported to reduce nonspecific protein fouling of PDMS.21–23 PEG can be introduced to PDMS surface by physical and chemical adsorption methods, direct covalent attachment, and graft polymerization. Pinto et al.24 reported that the PDMS surface can be treated with a plasma and then heated to introduce grafted PEG chains onto the surface. Tugulu et al.25 reported that PEG-methacrylate brush was polymerized on PDMS surface via surface-initiated atom transfer radical polymerization (ATRP). Ishihara et al.26–28 reported that PMPC graft chains can be introduced onto the PDMS surface. First, the PDMS surface is treated with a plasma, followed by application of the photo-radical initiator, benzophenone, to the PDMS surface to start graft polymerization of MPC. Some conventional graft polymerization methods require a long time to complete and can be complicated.
In this study, diblock copolymers (PMPC120-P(TSM/CEAx)y) composed of a hydrophilic PMPC block and random copolymer block with 3-(tris(trimethylsiloxy)silyl)propyl methacrylate (TSM) and 2-cinnamoylethyl acrylate (CEA) were prepared via reversible addition–fragmentation chain transfer (RAFT) radical polymerization.29 A thin film of PMPC120-P(TSM/CEAx)y was formed on the surface of the PDMS substrate using physical adsorption and photo-crosslinking (Fig. 1). The TSM units in the block copolymer can adsorb onto the PDMS.30 The pendant cinnamoyl group in the CEA units in PMPC120-P(TSM/CEAx)y can photo-crosslink due to photo dimerization of the cinnamoyl groups.31–34 Only the area that undergoes UV irradiation is selectively cross-linked. The polymer solution was applied to the PDMS to form thin polymer films, followed by exposure to UV light irradiation to improve the coating. The polymer chain was labeled with a fluorescent molecule at one end of the polymer chain to confirm adsorption onto the PDMS using a fluorescence microscope. Fluorescence-labeled bovine serum albumin (BSA) was used to evaluate protein antifouling of the polymer-coated PDMS. Cells were cultured on PMPC patterned film on PDMS for cellular patterning, because cells can adsorb onto the PDMS but not onto PMPC film.
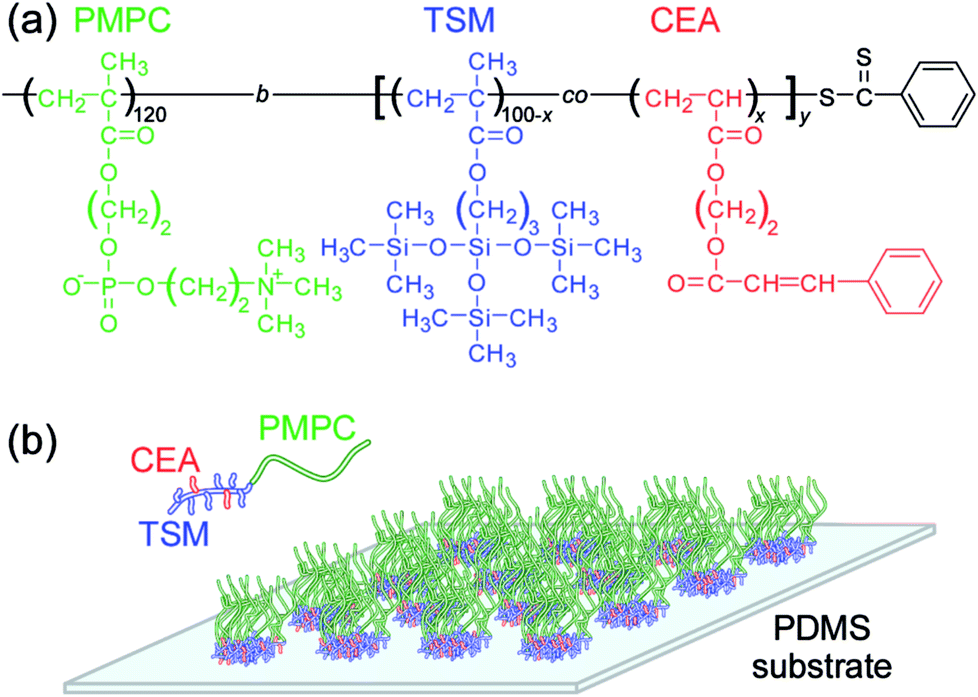 | ||
| Fig. 1 (a) Chemical structure of the diblock copolymer, PMPC120-P(TSM/CEAx)y. (b) Schematic illustration of pattern formation of PMPC120-P(TSM/CEAx)y onto the surface of the PDMS substrate. | ||
2. Experimental section
2.1 Materials
MPC was purchased from NOF Corp. (Japan), which was produced using a previously reported method.14 PMPC120 (Mn = 3.57 × 104 and Mw/Mn = 1.16) was prepared using a previously reported method.35 Detailed preparation method of PMPC120 was described in ESI.† 2-Cinnamoylethyl acrylate (CEA) was synthesized according to a previously reported method.36 3-(Tris(trimethylsiloxy)silyl)propyl methacrylate (TSM) was a gift from Shin-Etsu Chemical Co., Ltd. (Japan), which was passed through a disposable column (Aldrich) that removed any inhibitor. 4-Cyanopentanoic acid dithiobenzoate (CPD) was synthesized according to the method reported by Mitsukami and co-workers.37 4,4′-Azobis(4-cyanopentanoic acid) (V-501, 98%) and 4,4′-azobisisobutyronitrile (AIBN, 98%) were purchased from Wako Pure Chemical (Japan). Alexa Fluor 488 C5-maleimide from Invitrogen (U.S.), rhodamine 6G (99%) from Aldrich (U.S.), bovine serum albumin (BSA), Alexa Fluor 488 conjugate (488-BSA) from Molecular Probes (U.S.), fibronectin from BD Biosciences (Japan), and phosphate-buffered saline (PBS) from Gibco (U.S.) were used without further purification. Ethanol, methanol, and tetrahydrofuran (THF) were dried over 4 Å molecular sieves and purified by distillation. Water was purified with a Millipore Milli-Q system. The quartz substrate was 1 cm × 1 cm and 1 mm thick. The precursor of PDMS and crosslinker (Slipot 184 W/C, Dow Corning Toray) were fully mixed (10/1 = w/w). The mixture was evenly spread on a silicon plate and degassed in a vacuum for 2 h. The curing reaction was then conducted at 70 °C for 1 h. The PDMS substrate was cut into 1 cm × 1 cm pieces with a thickness of 1.5 mm. The L929 mouse fibroblast cells were provided by Riken BRC (Japan). The L929 cells were cultured in minimum essential medium (MEM, Sigma) supplemented with 10% (v/v) horse serum (Lonza), penicillin–streptomycin (100 units penicillin and 100 μg streptomycin per mL, Sigma), and 0.1 mM MEM non-essential amino acid (Gibco).2.2 Synthesis of PMPC120-PTSM68
PMPC120 (1.28 g, 0.036 mmol, Mn (NMR) = 3.57 × 104, Mw/Mn = 1.16), TSM (1.50 g, 3.55 mmol), and AIBN (5.89 mg, 0.036 mmol) were dissolved in ethanol (12 mL). The solution was deoxygenated with Ar gas for 30 min. The solution was stirred at 60 °C for 13 h. After polymerization, the solution was added to a large excess of THF to precipitate the polymer. The polymer was purified by reprecipitation from ethanol into a large excess of THF. The resulting polymer (PMPC120-PTSM68) was dried under vacuum at 40 °C overnight (1.71 g, 61.5%). Mn (NMR) for PMPC120-PTSM68 and DP for the PTSM block were 6.45 × 104 and 68, respectively, estimated from 1H NMR.2.3 Synthesis of PMPC120-P(TSM/CEA10)58
PMPC120 (599.6 mg, 0.017 mmol, Mn (NMR) = 3.57 × 104, Mw/Mn = 1.16), TSM (434.9 mg, 1.03 mmol), CEA (62.8 mg, 0.26 mmol), and AIBN (2.75 mg, 0.017 mmol) were dissolved in ethanol (4.2 mL). The solution was deoxygenated with Ar gas for 40 min. The solution was stirred at 60 °C for 20 h. After polymerization, the mixture was poured into a large excess of acetone. The precipitate was re-dissolved in methanol, which was poured into a large excess of acetone for purification by reprecipitation. The resulting polymer (PMPC120-P(TSM/CEA10)58) was dried under vacuum at 40 °C overnight (0.683 g, 62.2%). Mn (NMR) for PMPC120-P(TSM/CEA10)58, DP for the P(TSM/CEA10) block, and the content of CEA were 5.92 × 104, 58, and 10 mol%, respectively, estimated from 1H NMR. PMPC120-P(TSM/CEA9)33 was prepared by the same method described above. Mn (NMR) for PMPC120-P(TSM/CEA9)33, DP for the P(TSM/CEA9) block, and the content of CEA were 4.62 × 104, 33, and 9 mol%, respectively. Synthetic route for PMPC120-P(TSM/CEAx)y was shown in Fig. S1.†2.4 Fluorescent labeling at the polymer chain end
The polymer prepared via the RAFT process introduced a thiol group at the end of the polymer chain. The terminal dithioester group can be decomposed to a thiol group by aminolysis using reagents containing a primary amine group.38 The fluorescent molecule can be introduced at the terminal group of the polymer using a “thiol-ene” click reaction.39 A typical procedure for the fluorescence labelling at the polymer chain end: PMPC120-P(TSM/CEA10)58 (150.8 mg, 2.37 μmol, Mn (NMR) = 5.92 × 104), 2-hydroxyethylamine (0.72 mmol, 43.7 mmol), and Alexa Fluor 488 C5-maleimide (0.17 mg, 0.24 μmol) were dissolved in ethanol (5 mL). The solution was stirred for 20 h in the dark at room temperature under an Ar atmosphere. After reaction, the mixture was dialyzed against methanol for 4 days. The polymer solution was poured into a large excess of acetone. The resulting fluorescently labeled polymer PMPC120-P(TSM/CEA10)58F was under vacuum at 40 °C overnight (57.6 mg, 38.2%). The PMPC120 and PMPC120-PTSM68 were fluorescently labeled using a similar method to obtain PMPC120F and PMPC120-PTSM68F. Synthetic route of the fluorescently labeled polymers was shown in Fig. S2.†2.5 Photo-crosslinking of the polymers
Photo-dimerization of cinnamoyl groups was conducted using an Asahi Spectra MAX-300 instrument equipped with a 300 W Xe lamp and a 275 nm cutoff filter (LUX275) at 275 nm < λ < 385 nm. Light intensity was 11.3 mW cm−2 at 350 nm. The polymer thin film was prepared on a quartz substrate using a spin-coat technique. To monitor the photo-reaction of the cinnamoyl groups in the polymer film on the quartz substrate, changes in the absorption spectra of the polymer film were monitored.2.6 Surface coating of PDMS substrate using PMPC-containing polymers
The PDMS substrate was washed with ethanol. The polymers were dissolved in ethanol at a concentration (Cp) of 10 g L−1. The polymer solutions were spin-coated onto the PDMS substrate using a Mikasa Spin Coater MS-A150 (Fig. 2). Acceleration times were less than 2 s and total spin times were 30 s. The polymer solution was dropped 10 times in a row at the same position with 3 s intervals. Spin speeds ranged from 500 to 2500 rpm. The spin-coated polymer film was air-dried for 1 day at room temperature, normal pressure and humidity. UV irradiation was directed toward the polymer film on PDMS substrate through a photomask (lattice pattern with 150 μm and slit pattern with 150 μm width) for 15 min. After UV irradiation, the PDMS substrate was washed with water to remove non-crosslinked polymers. To confirm the presence of PMPC on the surface of the PDMS substrate, an aqueous solution of rhodamine 6G (0.01 g L−1) was dropped onto the PMPC pattered surface, which was washed with water to remove excess rhodamine 6G. The polymer-covered PDMS substrate was observed using fluorescence microscopy. | ||
| Fig. 2 Process for the surface treatment of PDMS substrates using PMPC120-P(TSM/CEAx)y and UV irradiation through a photomask. | ||
2.7 Protein antifouling
A PBS buffer solution (20 μL) of 488-BSA (0.01 g L−1) was dropped onto the surface of patterned polymer-coated PDMS substrate, which was allowed to stand for 2 h. The PDMS substrates were washed with water, and then observed using fluorescence microscopy. After the observations, the PDMS substrates were stained with rhodamine 6G, and were observed using fluorescence microscopy to confirm the presence of PMPC on the surface of the PDMS substrate.2.8 Measurements
1H NMR spectra were obtained using a Bruker DRX-500 spectrometer operating at 500 MHz. GPC measurements were obtained using a Tosoh DP-8020 pump and RI-8020 refractive index detector equipped with a Shodex GF-1G guard column and Shodex GF-7M HQ column (exclusion limit ∼ 107) operating at 40 °C under a flow rate of 0.6 mL min−1. Phosphate buffer (50 mM, pH 9) containing 10 vol% acetonitrile was used as the eluent. The values of Mn (GPC) and Mw/Mn were calibrated with standard sodium poly(styrenesulfonate) samples. UV-Vis absorption spectra were recorded using a Jasco V-530 UV/Vis spectrophotometer. Fluorescence micrographs were obtained using a Keyence Biorevo BZ-8000 equipped with a Nikon PlanFluor ELWD DM 20× NAO 0.45 objective lens (excitation/emission 480/510 and 540/605 nm). Laser scattering micrographs were obtained using a Keyence 3D laser microscope VK-8700 equipped with a Nikon CF IC EPI Plan 20× objective lens. Scanning electron microscope (SEM) images were obtained with a Keyence VE-9800 containing a thermal emission gun operating at 1.0 kV. Samples for SEM were cross sections of PMPC-patterned PDMS substrates, which were treated with Pt-sputter using a Sanyu Electron Quick Coater SC-701MK II. The surface morphologies of the films were observed with an atomic force microscope (AFM) operating in tapping mode using an instrument with a SII Instruments SPI4000 Probe Station controller at room temperature. The contact angles were measured using a sessile drop technique with a Kyowa Interface Science Drop Master 300 instrument. A 1.0 μL drop of pure water was placed carefully on dried portions of the PDMS substrate using a micro-syringe. The image of the drop was captured within 1 s of drop deposition to minimize error due to evaporation. The surface elemental composition was analyzed using X-ray photoelectron spectroscopy (XPS) with a Thermo Fischer Scientific Inc. ESCALab 250 spectrometer employing monochromatic AlK X-ray radiation (1486.6 eV). The XPS spectra were recorded at a take-off angle of 90°. Information ca. 5–6 nm depth from the surface was obtained under this condition. The system was operated at 15 kV and 200 W. Background removal was conducted using Thermo Fischer Scientific Inc. advantage analysis software. Attenuated total reflection-infrared (ATR-IR) spectra were obtained using a Jasco FT/IR-4200 spectrometer with a Jasco ATR-PRO450S base kit. Spectra in the IR region from 500 to 3500 cm−1 were collected over 32 scans with a spectral resolution of 1.0 cm−1.2.9 Cell culturing
The patterned PDMS substrate was incubated with 20 μg mL−1 fibronectin for 30 min at room temperature, followed by washing 5 times with PBS without drying. Then, the cells were seeded in the serum-containing culture medium onto the patterned PDMS substrate at 4 × 104 cells per cm2 and incubated for 30 h to acquire the image. During image acquisition, phase contrast images were acquired using an inverted microscope with an Olympus 10× 0.3NA plan objective lens and an Andor Technology, DU897 iXonEM EMCCD camera.3. Results and discussion
3.1 Characterization of the polymers
The unimodal GPC elution curve (Fig. S3†) for PMPC120 with narrow Mw/Mn (=1.15) indicated that the polymer possessed well-controlled structure. 1H NMR spectra were measured for PMPC120 in D2O at 20 °C (Fig. 3a). the Mn (NMR) and DP for PMPC120 were calculated from the area integral intensity ratio of the peaks of the pendant methylene protons in PMPC block at 3.7 ppm and the terminal phenyl protons in dithiobenzoate group at 7.5–8.0 ppm. | ||
| Fig. 3 1H NMR spectra for (a) PMPC120 in D2O at 20 °C and (b) PMPC120-P(TSM/CEA9)26 in methanol-d4 at 50 °C. | ||
The values of DP for the TSM block and Mn (NMR) for PMPC120-PTSM68 were estimated from the area integral intensity ratio of the peaks of the pendant methylene protons in PMPC block at 3.7 ppm and the pendant methyl protons in TSM unit at 0.2 ppm, respectively. The DP and contents of CEA for the P(TSM/CEAx)y block in PMPC120-P(TSM/CEA10)58 and PMPC120-P(TSM/CEA9)26 were calculated from the area integral intensity ratio of peaks of the pendant methylene protons in the PMPC block at 3.7 ppm and the pendant methine proton in the CEA unit at 6.6 ppm. The theoretical number-average molecular weight (Mn (theory)) was calculated from the following eqn (1):
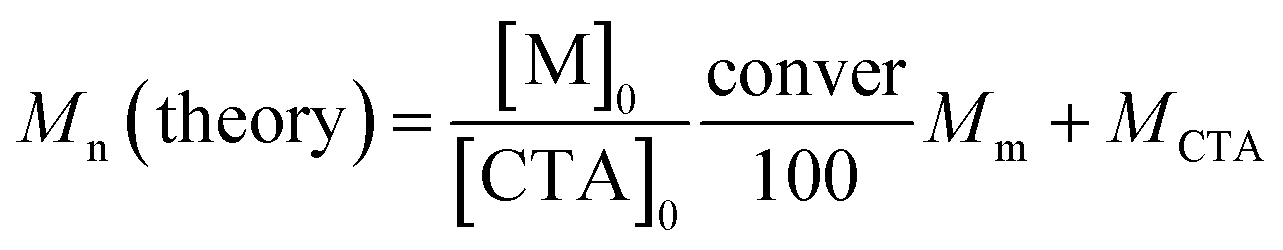 | (1) |
| Samples | Mn (theory)a × 104 | Mn (NMR)a × 104 | DPb | CEA contentc (mol%) |
|---|---|---|---|---|
| a Calculated from eqn (1).b Degree of polymerization estimated from NMR.c CEA content in the P(TSM/CEAx)y block.d Degree of polymerization of PTSM block.e Degree of polymerization of P(TSM/CEAx)y block. | ||||
| PMPC120 | 3.57 | 3.57 | 120 | — |
| PMPC120-PTSM68 | 6.87 | 6.45 | 68d | — |
| PMPC120-P(TSM/CEA9)26 | 4.97 | 4.62 | 26e | 9 |
| PMPC120-P(TSM/CEA10)58 | 6.37 | 5.92 | 58e | 10 |
3.2 Photo-crosslinking conditions
The ethanol solution of PMPC120-P(TSM/CEA10)58 was spin-coated onto the quartz glass surface to prepare a thin polymer film. An absorption peak with a maximum wavelength of 275 nm attributed to the pendant cinnamoyl groups in PMPC120-P(TSM/CEA10)58 was observed in the UV-Vis absorption spectra. Absorption spectral changes for the polymer thin film upon UV irradiation for varying times were studied (Fig. S4†). Absorbance decreased with increasing UV irradiation time. This decrease in absorbance was caused by trans-to-cis photoisomerization and 2 + 2 photocycloaddition reactions of the cinnamoyl groups.40 Absorbance of the cinnamoyl groups decreased to 76% after 15 min UV irradiation.3.3 Coating of PMPC-containing polymers onto the PDMS substrate
An ethanol solution of fluorescently labeled PMPC120-P(TSM/CEA10)58F was dropped onto the surface of PDMS to prepare a thin film by spin coating, followed by irradiation with UV light through a lattice-patterned photomask for 15 min. After washing with water, the patterned PDMS substrate was observed using fluorescence microscopy (Fig. 4a). The green fluorescence attributed to PMPC120-P(TSM/CEA10)58F allowed observation of the pattern of the photomask. The polymer thin film without photo-crosslinking was prepared by spin-coating on the surface of the PDMS substrate, which was easily rinsed off from the substrate using water. After UV irradiation, the molecular weight of the hydrophobic portion of the diblock copolymer increased due to crosslinking of the pendant cinnamoyl groups, which made the polymer thin film insoluble in water. The area of the polymer thin film that was not irradiated (due to shading by the photomask) was easily rinsed off by washing with water. The thickness of the polymer film on the PDMS substrate was measured with a scanning laser microscope (Fig. S5†). Typical thickness of the polymer thin film was ca. 260 nm. | ||
| Fig. 4 Fluorescence micrographs of (a) PMPC120-P(TSM/CEA10)58F coated on the surface of the PDMS substrate and (b) PMPC120-P(TSM/CEA10)58F coated on the surface of a PDMS substrate stained with rhodamine 6G. | ||
Rhodamine 6G is a cationic and lipophilic dye, which adsorbs specifically to phosphatidylcholine. The rhodamine 6G dye can adsorb to phospholipid-based polymers, including PMPC.41 An aqueous rhodamine 6G solution was dropped onto a patterned PDMS substrate to stain PMPC. After washing with water to remove excess dye, the PDMS substrate was observed using fluorescence microscopy (Fig. 4b). The presence of PMPC in a square lattice pattern reflected the photomask, because the red fluorescence attributed to rhodamine 6G can be observed in the mask pattern. The square area with red fluorescence from rhodamine 6G was completely overlapped with the area of green fluorescence from PMPC120-P(TSM/CEA10)58F. These data indicate that only the UV-light irradiated area can be coated selectively by biocompatible PMPC on the surface of a PDMS substrate.
The diblock copolymer can adsorb weakly to the PDMS surface because of the affinity of the P(TSM/CEAx)y block and PDMS substrate. The polymer can form a water-insoluble patterned film due to photo-crosslinking by UV irradiation, because the molecular weight of the hydrophobic portion of the polymer was increased. The diblock copolymers were expected to dissolve in water and so be removed easily from the PDMS substrate when the polymers did not crosslink. To confirm this hypothesis, the following study was performed. Ethanol solutions of PMPC120F and PMPC120-PTSM68F, which do not contain photo-crosslinking groups in their chemical structures, were dropped onto a PDMS substrate to prepare a thin film by spin coating. The lattice patterned photomask was exposed to UV light for 15 min. After washing with water, the PDMS substrates were observed using fluorescence microscopy (Fig. S6†). The patterned green fluorescence from the polymer films on the PDMS substrate were not observed, which indicates that polymer films without photo-crosslinking were rinsed off easily from the PDMS substrate by washing with water.
SEM images for PMPC120-P(TSM/CEA10)58-patterned film on a PDMS substrate and its cross section were measured (Fig. S7†). A clear lattice pattern from the photomask on the PDMS substrate could be observed, suggesting that a polymer-patterned film can be formed upon UV irradiation. Polymer film thickness was ca. 290 nm, as estimated from the cross-sectional SEM image, which was close to the value (ca. 260 nm) estimated from the scanning laser microscope observations. AFM measurements were performed for PMPC120-P(TSM/CEA10)58-patterned film on a PDMS substrate to observe the surface topography (Fig. S8†). Typical thickness of the polymer thin film was ca. 190 nm estimated from AFM.
The contact angles of pure water were measured on the PDMS substrate with and without the polymer thin film (Fig. 5). Contact angles of the PDMS substrates without and with the polymer film were 107.9° and 37.5°, respectively. The surface of bare PDMS substrate has a hydrophobic nature. The PMPC120-P(TSM/CEA10)58-covered PDMS substrate possessed a highly wettable surface, because the PDMS surface was covered with the hydrophilic PMPC block.
 | ||
| Fig. 5 Water contact angles of the surface of the PDMS substrate (a) without and (b) with a coating of PMPC120-b-P(TSM/CEA10)58. | ||
To confirm the surface coverage of PMPC120-P(TSM/CEA10)58 on the PDMS substrate, XPS studies were conducted for bare and polymer-coated PDMS substrates (Fig. 6). The PMPC120-P(TSM/CEA10)58 film on the PDMS substrate was prepared using UV irradiation without the photomask. The photoelectron peaks of the oxygen, carbon, and silicon energy levels were observed in the survey spectrum of bare PDMS substrate (Fig. 6a).42 The presence of nitrogen and phosphorous could not be confirmed by the narrow scans used for bare PDMS substrate. For the PMPC120-P(TSM/CEA10)58-coated PDMS substrate, peaks for phosphorus P2p at 128 eV and nitrogen N1s at 399 eV were observed in the narrow XPS spectra. These peaks were attributed to the pendant phosphorylcholine groups in PMPC120-P(TSM/CEA10)58.43,44 This observation indicates that PMPC120-P(TSM/CEA10)58 was coated on the surface of the PDMS substrate. The C1s spectrum of PMPC120-P(TSM/CEA10)58-coated PDMS can be resolved into C–C, C–O, and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O. All these peaks can be assigned to the polymer structure (Fig. S9†). The XPS surface elemental composition for bare PDMS was agreed well with the calculated composition (Table S1†). On the other hand, the XPS elemental composition for PMPC120-P(TSM/CEA10)58-coated PDMS was not agreed with the calculated composition. The compositions of phosphorous and nitrogen estimated from XPS were smaller than calculated values, and the XPS composition of silicon was larger than calculated value. These observations suggest that some hydrophobic TSM units in the block copolymer may localize on solid–gas interface of the polymer film due to hydrophobicity of air.
O. All these peaks can be assigned to the polymer structure (Fig. S9†). The XPS surface elemental composition for bare PDMS was agreed well with the calculated composition (Table S1†). On the other hand, the XPS elemental composition for PMPC120-P(TSM/CEA10)58-coated PDMS was not agreed with the calculated composition. The compositions of phosphorous and nitrogen estimated from XPS were smaller than calculated values, and the XPS composition of silicon was larger than calculated value. These observations suggest that some hydrophobic TSM units in the block copolymer may localize on solid–gas interface of the polymer film due to hydrophobicity of air.
 | ||
| Fig. 6 XPS survey spectra of (a) a bare PDMS substrate and (b) PMPC120-P(TSM/CEA10)58-coated PDMS substrate. XPS narrow spectra for (c) P2p and (d) N1s for PMPC120-P(TSM/CEA10)58-coated PDMS substrate. | ||
The coverage of the polymer film on the PDMS substrate was confirmed using ATR-FTIR measurements. ATR-FTIR spectra were measured for bare PDMS substrate, PMPC120-P(TSM/CEA10)58 powder, and PMPC120-P(TSM/CEA10)58-covered PDMS substrate (Fig. 7). For the bare PDMS substrate, two absorption bands at 1070 and 790 cm−1 were attributed to the Si–Oasym, and the absorption band at 1260 cm−1 was attributed to CH3sym groups, while absorption bands at 2960 cm−1 corresponded to asymmetric and symmetric stretching of the CH3 groups (Fig. 7a).45 In the spectrum of the PMPC120-P(TSM/CEA10)58-coated PDMS substrate, characteristic absorption bands at 2900 cm−1 (C–H) and 1728 cm−1 (ester, CO) were observed, which were characteristic of PMPC120-P(TSM/CEA10)58 (Fig. 7c).46 This observation indicates that the coating of PMPC120-P(TSM/CEA10)58 on the PDMS substrate was achieved successfully.
 | ||
| Fig. 7 ATR-IR spectra for (a) bare PDMS substrate, (b) PMPC120-P(TSM/CEA10)58 powder, and (c) PDMS substrate coated with PMPC120-P(TSM/CEA10)58. | ||
3.4 Protein antifouling effect of the polymer-coated PDMS substrate
The square areas were irradiated with UV light to fix PMPC through crosslinking of the block copolymers (Fig. 8a). PMPC fixed in the square areas inhibited the adsorption of 488-BSA. Non-crosslinked PMPC120-P(TSM/CEA10)58 was rinsed off from the non-irradiated area. Therefore, green fluorescence from 488-BSA was observed from non-irradiated area, suggesting that 488-BSA adsorbed there. After confirmation of protein antifouling on the surface of the PDMS substrate, an aqueous rhodamine 6G solution was dropped onto the substrate (Fig. 8b). Red fluorescence emission from rhodamine 6G can be observed in the square areas where PMPC was fixed. | ||
| Fig. 8 Fluorescence micrographs of (a) PMPC120-P(TSM/CEA10)58 coated on the surface of PDMS substrate after adsorption of 488-BSA and (b) PMPC120-P(TSM/CEA10)58 coated on PDMS substrate stained with rhodamine 6G. | ||
A 488-BSA buffer solution was dropped onto the surface of the PMPC120-P(TSM/CEA9)26-coated PDMS substrate, followed by observation with fluorescence microscopy after washing with PBS buffer (Fig. S10†). Green fluorescence of 488-BSA was observed from the polymer-coated area, suggesting that protein antifouling efficiency was low. Interaction of the PDMS substrate with PMPC120-P(TSM/CEA9)26 was weaker than that with PMPC120-P(TSM/CEA10)58, because the P(TSM/CEAx)y block in PMPC120-P(TSM/CEA9)26 is shorter than that in PMPC120-P(TSM/CEA10)58. Therefore, PMPC120-P(TSM/CEA9)26 was desorbed from the PDMS substrate, even after photo-crosslinking of the pendant cinnamoyl groups.
3.5 Cell patterning
To test the effectiveness of protein antifouling by PMPC on cell patterning, the behavior of living L929 mouse fibroblasts on PMPC-patterned PDMS substrates was investigated by microscopy. The typical behavior of cells on the PMPC-patterned PDMS substrate was shown in Fig. 9 with lattice pattern and Fig. S11† with slit pattern. Within 1 h after seeding, the cells outside the PMPC area started to spread, followed by spreading and growth of the cells on the area with no PMPC coating. The cells proliferated and actively migrated on the substrate. However, cell migration was confined almost exclusively to the area without PMPC coating, therefore, the cells formed a pattern that was lattice shaped. Even after 30 h incubation, cells migrated only within the area without PMPC coating, suggesting stable and long-term suppression of protein absorption by PMPC and resulting cell patterning effectiveness. | ||
| Fig. 9 Typical behavior of L929 cells on a lattice PMPC-patterned PDMS substrate. | ||
4. Conclusions
The pendant photo-crosslinkable groups containing amphiphilic diblock copolymers, PMPC120-P(TSM/CEAx)y, were prepared via RAFT controlled/living radical polymerization using PMPC120 macro-CTA. PMPC was patterned onto the PDMS substrate using UV irradiation with a photomask and PMPC120-P(TSM/CEAx)y. The diblock copolymer was used to coat the surface of the microchannels to avoid adsorption of proteins and allow cell patterning. The micro fluidic channels could be covered easily with PMPC. The polymer solution was added to the micro fluidic channels made from PDMS, followed by UV irradiation to coat the inside wall of the channels, because the PDMS surface can be coated by PMPC120-P(TSM/CEAx)y upon the irradiation.Acknowledgements
This work was financially supported by a Grant-in-Aid for Scientific Research (25288101) from the Japan Society for the Promotion of Science (JSPS), and the Cooperative Research Program, Network Joint Research Center for Materials and Devices (2013B25). The L929 mouse fibroblasts were provided by Riken BRC through the National Bio-Resource Project of the MEXT, Japan.Notes and references
- C. H. Seo, H. Jeong, K. S. Furukawa, Y. Suzuki and T. Ushida, Biomaterials, 2013, 34, 1764–1771 CrossRef CAS PubMed.
- M. Chhabra, J. M. Prausnitz and C. J. Radke, Biomaterials, 2007, 28, 4331–4342 CrossRef CAS PubMed.
- M. K. Horne III and K. J. Brokaw, Thromb. Res., 2003, 112, 111–115 CrossRef PubMed.
- T. J. Joyce and A. Unworth, Wear, 2001, 250, 199–205 CrossRef.
- H. Miyoshi and T. Adachi, Tissue Eng., Part B, 2014, 20, 609–627 CrossRef CAS PubMed.
- D. Falconnet, G. Csucs, H. M. Grandin and M. Textor, Biomaterials, 2006, 27, 3044–3063 CrossRef CAS PubMed.
- J. El-Ali, P. K. Sorger and K. F. Jensen, Nature, 2006, 442, 403–411 CrossRef CAS PubMed.
- J. Doh and D. J. Irvine, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5700–5705 CrossRef CAS PubMed.
- C. M. Nelson, R. P. Jean, J. L. Tan, W. F. Liu, N. J. Sniadecki, A. A. Spector and C. S. Chen, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 11594–11599 CrossRef CAS PubMed.
- W. F. Liu, C. M. Nelson, D. M. Pirone and C. S. Chen, J. Cell Biol., 2006, 173, 431–441 CrossRef CAS PubMed.
- S. N. Bhatia, U. J. Balis, M. L. Yarmush and M. Toner, FASEB J., 1999, 13, 1883–1900 CAS.
- C. Rodriguez-Emmenegger, C. M. Preuss, B. Yameen, O. Pop-Georgievski, M. Bachmann, J. O. Mueller, M. Bruns, A. S. Goldmann, M. Bastmeyer and C. Barner-Kowollik, Adv. Mater., 2013, 25, 6123–6127 CrossRef CAS PubMed.
- A. F. Hirschbiel, S. Geyer, B. Yameen, A. Welle, P. Nikolov, S. Giselbrecht, S. Scholpp, G. Delaittre and C. Barner-Kowollik, Adv. Mater., 2015, 27, 2621–2626 CrossRef CAS PubMed.
- K. Ishihara, T. Ueda and N. Nakabayashi, Polym. J., 1990, 22, 355–360 CrossRef CAS.
- T. Ueda, H. Oshida, K. Kurita, K. Ishihara and N. Nakabayashi, Polym. J., 1992, 24, 1259–1269 CrossRef CAS.
- M. Kyomoto, T. Moro, S. Yamane, K. Watanabe, M. Hashimoto, Y. Takatori, S. Tanaka and K. Ishihara, Biomaterials, 2014, 35, 6677–6686 CrossRef CAS PubMed.
- M. Kyomoto, Y. Iwasaki, T. Moro, K. Saiga, F. Miyaji, H. Kawaguchi, Y. Takatori, K. Nakamura and K. Ishihara, Biomaterials, 2010, 31, 658–668 CrossRef CAS PubMed.
- M. Kyomoto, T. Moro, S. Yamane, M. Hashimoto, Y. Takatori and K. Ishihara, Biomaterials, 2013, 34, 7829–7839 CrossRef CAS PubMed.
- T. Moro, Y. Takatori, K. Ishihara, T. Konno, Y. Takigawa, T. Matsushita, U.-I. Chung, K. Nakamura and H. Kawaguchi, Nat. Mater., 2004, 3, 829–836 CrossRef CAS PubMed.
- T. Talarico, A. Swank and C. Privalle, Biochem. Biophys. Res. Commun., 1998, 250, 354–358 CrossRef CAS PubMed.
- C. Blaszykowski, S. Sheikh and M. Thompson, Chem. Soc. Rev., 2012, 41, 5599–5612 RSC.
- C. J. Fristrup, K. Jankova and S. Hvilsted, Soft Matter, 2009, 5, 4623–4634 RSC.
- Y. Iwasaki, M. Takamiya, R. Iwata, S. Yusa and K. Akiyoshi, Colloid. Surf., B, 2004, 39, 133–142 CrossRef PubMed.
- S. Pinto, P. Alves, C. M. Matos, A. C. Santos, L. R. Rodrigues, J. A. Teixeira and M. H. Gil, Colloids Surf., B, 2010, 81, 20–26 CrossRef CAS PubMed.
- S. Tugulu and H. A. Klok, Macromol. Symp., 2009, 279, 103–109 CrossRef CAS PubMed.
- T. Goda, T. Konno, M. Takai, T. Moro and K. Ishihara, Biomaterials, 2006, 27, 5151–5160 CrossRef CAS PubMed.
- T. Goda, R. Matsuno, M. Takai and K. Ishihara, Colloids Surf., B, 2008, 63, 64–72 CrossRef CAS PubMed.
- T. Goda and K. Ishihara, Expert Rev. Med. Devices, 2006, 3, 167–174 CrossRef CAS PubMed.
- J. Chifari, Y. K. Chong, F. Ercole, J. Krstina, T. P. Le, R. T. A. Mayadunne, G. F. Meijs, G. Moad, C. L. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef.
- G. Schreyeck and P. Marie, Langmuir, 1999, 15, 8212–8219 CrossRef CAS.
- M. Kato, T. Ichijo, K. Ishii and M. Hasegawa, J. Polym. Sci., Part A: Polym. Chem., 1971, 9, 2109–2128 CrossRef CAS PubMed.
- A. Guo, G. Liu and J. Tao, Macromolecules, 1996, 29, 2487–2493 CrossRef CAS.
- S. Murase, K. Kinoshita, K. Horie and S. Morino, Macromolecules, 1997, 30, 8088–8090 CrossRef CAS.
- G. Wiegand, T. Jaworek, G. Wegner and E. Sackmann, Langmuir, 1997, 13, 3563–3569 CrossRef CAS.
- S. Yusa, K. Fukuda, T. Yamamoto, K. Ishihara and Y. Morishima, Biomacromolecules, 2005, 6, 663–670 CrossRef CAS PubMed.
- S. Yusa, M. Sugahara, T. Endo and Y. Morishima, Langmuir, 2009, 25, 5258–5265 CrossRef CAS PubMed.
- Y. Mitsukami, M. S. Donovan, A. B. Lowe and C. L. McCormick, Macromolecules, 2001, 34, 2248–2256 CrossRef CAS.
- R. T. A. Mayadunne, E. Rizzardo, J. Chiefari, J. Krstina, G. Moad, A. Postma and S. H. Thang, Macromolecules, 2000, 33, 243–245 CrossRef CAS.
- J. Akimoto, M. Nakayama, K. Sakai and T. Okano, Biomacromolecules, 2009, 10, 1331–1336 CrossRef CAS PubMed.
- Y. Iwasaki, A. Matsumoto and S. Yusa, ACS Appl. Mater. Interfaces, 2012, 4, 3254–3260 CAS.
- J. H. Wang, J. D. Bartlett, A. C. Dunn, S. Small, S. L. Willis, M. J. Driver and A. L. Lewis, J. Microsc., 2005, 217, 216–224 CrossRef CAS PubMed.
- L. Yang, L. Li, Q. Tu, L. Ren, Y. Zhang, X. Wang, Z. Zhang, W. Liu, L. Xin and J. Wang, Anal. Chem., 2010, 82, 6430–6439 CrossRef CAS PubMed.
- K. Futamura, R. Matsuno, T. Konno, M. Takai and K. Ishihara, Langmuir, 2008, 24, 10340–10344 CrossRef CAS PubMed.
- Y. Iwasaki, U. Takami, Y. Shinohara, K. Kurita and K. Akiyoshi, Biomacromolecules, 2007, 8, 2788–2794 CrossRef CAS PubMed.
- E. J. Park, Y. K. Cho, D. H. Kim, M. G. Jeong, Y. H. Kim and Y. D. Kim, Langmuir, 2014, 30, 10256–10262 CrossRef CAS PubMed.
- P. Akkahat, S. Kiatkamjornwong, S. Yusa, V. P. Hoven and Y. Iwasaki, Langmuir, 2012, 28, 5872–5881 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional polymerization method, reaction schemes, GPC, UV absorption, laser micrograph, fluorescence microscope, SEM, AFM, and XPX data. See DOI: 10.1039/c5ra08843g |
| This journal is © The Royal Society of Chemistry 2015 |