
Amine-catalyzed cascade reactions of ketoses with 1,3-dicarbonyl compounds†
Abstract
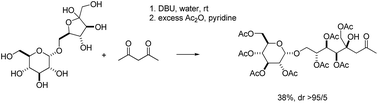
An amine-catalyzed cascade reaction of ketoses with 1,3-dicarbonyl compounds is described. Several highly chemo- as well as stereoselective reactions are operating in this novel cascade. The operationally simple protocol allows stereoselective access to optically active carbon chain elongated ketoses.
 Please wait while we load your content...
Please wait while we load your content...

