
Influence of the HDPE molecular weight and content on the morphology and properties of the impact polypropylene copolymer/HDPE blends†
Abstract
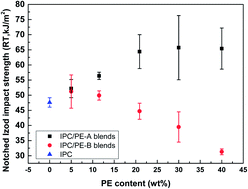
Two HDPE resins with different molecular weights were blended with one impact polypropylene copolymer with varied weight contents. The influence of molecular weight and content of PE on the morphology and properties of the blends was studied. It is found that by changing the molecular weight and weight content of PE, the morphologies of the blends show great differences. Spherical particles with different structure details and obvious orientation structures were found. Properties, such as impact strength and elongation at break, depend not only the composition but also on the morphology. This study may shed light on the structure–property relationship study in polyolefin impact blends.
 Please wait while we load your content...
Please wait while we load your content...

