
Solvothermal synthesis of MoS2 nanospheres in DMF–water mixed solvents and their catalytic activity in hydrocracking of diphenylmethane†
Hui Dua,
Dong Liu*a,
Min Lia,
Raja L. Al Otaibi*b,
Renqing Lvc and
Yadong Zhanga
aState Key Laboratory of Heavy Oil Processing, China University of Petroleum, Qingdao 266555, Shandong, P. R. China. E-mail: ldongupc@vip.sina.com; Tel: +86-532-86984629
bPetrochemical Research Institute, King Abdulaziz City for Science and Technology, P.O. Box 6086, Riyadh 11442, Kingdom of Saudi Arabia. E-mail: raletabi@kacst.edu.sa
cCollege of Science, China University of Petroleum, Qingdao 266555, Shandong, P. R. China
First published on 8th September 2015
Abstract
MoS2 nanospheres were successfully prepared via a solvothermal process using 1-ethyl-3-methylimidazolium bromide ([EMIM]Br) in a mixed solvent of DMF–water. A probable [EMIM]Br aggregation in different mixed solvents and the formation mechanism of the MoS2 nanospheres are presented. The MoS2 nanospheres delivered high catalytic activity in hydrocracking of diphenylmethane.
Molybdenum disulfide (MoS2) with a layered and hexagonal structure is used as a catalyst,1 solid lubricant,2 electrode,3 and in hydrogen storage media.4 As a catalyst, the layered anisotropic structure of MoS2 provides a massive “edge area” at which the catalytic interaction of reactants with MoS2 edges and defects occurs.5,6 During recent decades, a variety of catalysts based on MoS2 have been used in processes such as the hydrocracking and hydrodesulfurization of petroleum,7,8 the hydrodeoxygenation of bio-oil,9 and the production of chemicals.10,11 Compared with supported catalysts, unsupported catalysts have the advantages of high dispersion and avoidance of pore-plugging in the residue hydrocracking process. As a consequence, dispersed MoS2 catalysts have been extensively employed in ENI-EST, SOC, and (HC)3 CASH hydrorefining technologies of residue.12–14
MoS2 with different micro/nano structures has been synthesized by multiple methods, such as a solvo-hydrothermal method,15–17 sonochemical method,18,19 chemical vapor deposition,20,21 a chemical solution route,22 microwave synthesis,23 and a thermal sulfurization method.24 The solvo-hydrothermal approach is an effective and simple way to prepare micro/nanostructured materials at low temperature.25 In recent years, ionic liquids (ILs) have been used as templates in hydrothermal synthesis of various micro/nano-structured inorganic materials.16,26,27 Ma et al. obtained MoS2 microspheres with average diameter about 2.1 μm via a hydrothermal method assisted by [BMIM][BF4].16 Hollow MoS2 microspheres with diameters ranging from 1.8 μm to 2.1 μm were prepared in [BMIM]Cl/water emulsions.28 In the same ionic liquids/water medium, hollow vesicle-like MoS2 microspheres (1–2 μm) were synthesized with (NH4)2MoS4 as precursor and hydrazine hydrate as reductant.29 However, to our knowledge, there are no reports on preparation of MoS2 nanospheres by a solvo-hydrothermal route assisted by ionic liquid.
In this research, we present a solvothermal route for synthesis of MoS2 nanospheres using a small molecule ionic liquid (1-ethyl-3-methylimidazolium bromide, [EMIM]Br) as a template in dimethyl formamide (DMF)–water mixed solvents. A probable [EMIM]Br aggregation in different mixed solvents and the formation mechanism of MoS2 nanospheres are presented. Additionally, the catalytic activity of the as-prepared MoS2 products was investigated in diphenylmethane hydrocracking.
Detailed experiments from the ionic liquid-assisted solvothermal synthesis of MoS2 products in DMF–water mixed solvents to their catalytic activity in hydrocracking of diphenylmethane are described in the ESI.† XRD patterns of the as-prepared MoS2 products are demonstrated in Fig. 1. The major diffraction peaks can be indexed to the hexagonal 2H MoS2 (JCPDS 37-1492). As shown in Fig. 1a–c, the absence of a relatively strong diffraction peak at around 2θ = 14° (002) reveals that the well-stacked layered structure of MoS2 did not form during the solvothermal process. Meanwhile, the diffraction peaks at 2θ = 8.6° and 17.6° are considered to be the (002) and (004) peaks shifting to lower angle range, which has been confirmed by other researchers.17,29 This is a consequence of the enlarged interlayer spacing caused by the ILs between MoS2 layers. Compared with Fig. 1a, the weak peaks in Fig. 1b and c show that the crystallinity of MoS2 products becomes poor in the presence of DMF. After annealing at 700 °C for 2 h in the mixed atmosphere of N2/H2 (9/1, v/v), the MoS2 products exhibit a strong diffraction peak at 2θ = 14.2° (Fig. 1d), implying that the crystallinity of MoS2 products is improved by annealing.
 | ||
| Fig. 1 XRD patterns of MoS2 samples synthesized at 200 °C for 24 h. (a) V1/V2 (DMF/water) = 0/9; (b) V1/V2 = 3/6; (c) V1/V2 = 5/4; and (d) after annealing of (b) in the mixed atmosphere of N2/H2 (9/1, v/v) at 700 °C for 2 h. *Shifted peaks of (002) and (004). | ||
The morphology and structure of MoS2 nanospheres, synthesized in mixed solvents of V1/V2 = 3/6, before and after annealing were characterized by SEM and TEM. Fig. 2a and b shows that the MoS2 products are nanospheres with a mean diameter of 400 nm. The MoS2 nanospheres with rough surface are made up of numerous petal-shaped nanoflakes, as can be seen from the clear view of a MoS2 nanosphere in Fig. 2a. From the BET measurement, the specific surface area of MoS2 nanospheres is 67.9 m2 g−1. In addition, the HRTEM image (Fig. 2c) clearly reveals that the MoS2 nanoflakes were poorly stacked with fewer layers (3–8 layers) and that the average spacing of MoS2 layers is about 0.91 nm. ILs, inside MoS2 nanospheres and between MoS2 layers, would vanish during annealing at 700 °C. Therefore, the MoS2 nanospheres become irregular in shape, and the size decreases to 300–400 nm (as shown in Fig. 2d and e). Fig. 2f shows that the petal-shaped MoS2 nanoflakes were straightened by annealing, and that the number of layers in each flake increases to dozens with a characteristic MoS2 layer spacing of 0.65 nm (002). The HRTEM results are consistent with the XRD patterns.
 | ||
| Fig. 2 SEM, TEM and HRTEM images of MoS2 samples synthesized at 200 °C for 24 h in V1/V2 = 3/6. (a–c) before annealing, (d–f) after annealing in the mixed atmosphere of N2/H2 (9/1, v/v) at 700 °C for 2 h. | ||
The ratio of DMF to water has a significant influence on the morphology of MoS2 products. As shown in Fig. 3a, hollow MoS2 microspheres with diameter of 1.7–3.8 μm were synthesized in the absence of DMF. However, irregular and agglomerated MoS2 nanoflakes were obtained in the mixed solvents of V1/V2 = 5/4 (Fig. 3c). Compared with the nanoflakes in MoS2 nanospheres (Fig. 2c), the shape of these MoS2 nanoflakes (Fig. 3d) was straighter during the solvo-hydrothermal process. The specific surface areas of the hollow MoS2 microspheres and MoS2 nanoflakes are 35.1 m2 g−1 and 49.7 m2 g−1, respectively. Both the products in Fig. 3 were poorly stacked with the average spacing of MoS2 layers being greater than 0.9 nm. The full width of half maximum (FWHM) values of the main peaks of MoS2 samples synthesized in different solvents are listed in Table 1. The FWHM values of (002) and (004) reflections increase with increasing DMF content of mixed solvents. The smallest FWHM value implies the largest crystalline grain of MoS2 samples synthesized without DMF. As can be seen from the HRTEM images of MoS2 samples, the flakes in MoS2 samples synthesized without DMF have a more straight structure and MoS2 layers.
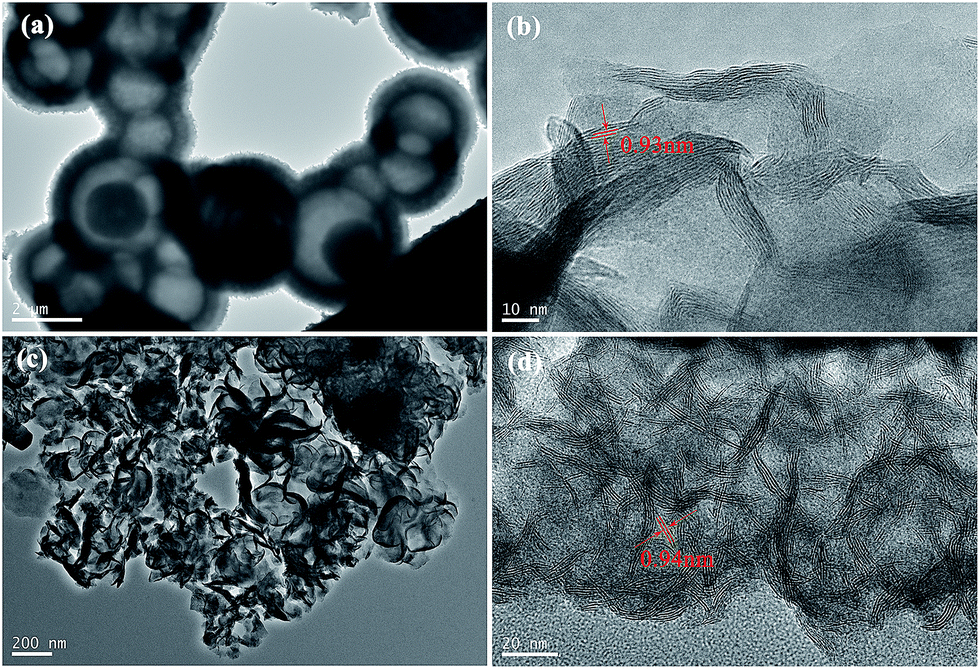 | ||
| Fig. 3 TEM and HRTEM images of as-prepared MoS2 samples synthesized in V1/V2 (DMF/water) = 0/9 (a and b), V1/V2 = 5/4 (c and d). | ||
| V1/V2 (DMF/water) | FWHM | |
|---|---|---|
| (002) | (004) | |
| 0/9 | 0.47° | 0.40° |
| 3/6 | 0.94° | 1.16° |
| 5/4 | 1.87° | 2.38° |
According to the literature,22 the reaction involved in the solvothermal synthesis process is as follows:
| 4(NH4)6Mo7O24 + 63CS(NH2)2 + 136HCl + 58H2O → 28MoS2 + 7(NH4)2SO4 + 136NH4Cl + 63CO2 |
It has been reported that ILs act as a template in solvo-hydrothermal synthesis of MoS2.16,28,29 [EMIM]+ can form vesicles in certain solvents under appropriate conditions. Then, Mo7O246− anions can adsorb onto the vesicle surfaces by electrostatic attraction,30 and react with the H2S in situ produced by hydrolysis of CS(NH2)2. Based on the experimental results, the probable IL aggregation in different solvents and the formation mechanism of MoS2 nanospheres synthesized in V1/V2 = 3/6 is presented in Scheme 1. ILs could form multilamellar vesicles with diameters of a few micrometers in pure water. With gradual addition of DMF, the multilamellar vesicles break because of higher energy, then small multilamellar vesicles or unilamellar vesicles are formed to obtain a low energy state.31 MoS2 nanospheres will be generated gradually on the surface of IL vesicles (V1/V2 = 3/6). However, if the addition of DMF continues, the stable aggregation structure of ILs could not form in the mixed solution (V1/V2 = 5/4).
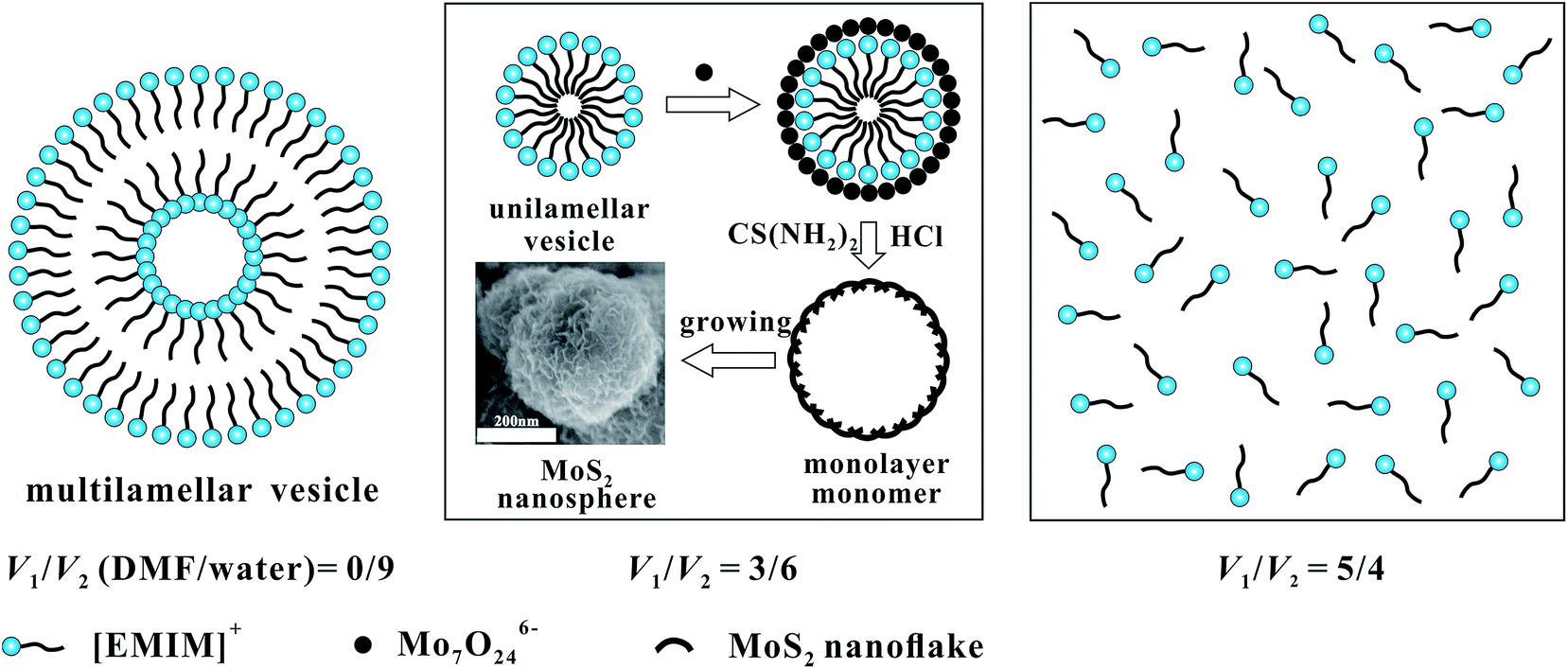 | ||
| Scheme 1 Schematic illustration of IL aggregation in different solvents and the formation mechanism of MoS2 nanospheres synthesized in V1/V2 = 3/6. | ||
Table 2 shows the results of diphenylmethane (DPM) hydrocracking in the presence of different MoS2 products at various temperatures. It can be seen that DPM barely hydrocracked when the reaction was performed at 450 °C without a catalyst, while all of the MoS2 products significantly facilitate conversion of DPM. Under the hydrocracking conditions, coordinatively unsaturated sites can be formed readily at the edge and corner positions of MoS2, which are the sites for hydrogen activation.32 Hydrogen molecules split to yield hydrogen free-radicals on the coordinatively unsaturated sites, then the produced hydrogen free-radicals add to the ipso position of DPM to trigger scission of the Car–Calk bond.33 The produced hydrogen free-radicals also can help to saturate the aromatic rings of DPM. Benzene and toluene are the main products of DPM hydrocracking, while small amounts of benzylcyclohexane and dicyclohexylmethane are also identified in the hydrocracked products.
| Catalyst | Temperature (°C) | Conversion of DPM (%) | Selectivity (mol%) | |||
|---|---|---|---|---|---|---|
| Benzene | Toluene | BCH | DHM | |||
| a BCH, benzylcyclohexane; DHM, dicyclohexylmethane. | ||||||
| None | 450 | 0.7 | 100 | 99.9 | 0 | 0 |
| MoS2 microsphere | 350 | 15.1 | 75.3 | 75.3 | 23.8 | 0.9 |
| 400 | 39.6 | 88.6 | 88.6 | 9.7 | 1.7 | |
| 450 | 52.5 | 90.1 | 90.1 | 7.3 | 2.6 | |
| MoS2 nanosphere | 350 | 18.9 | 69.5 | 69.5 | 29.4 | 1.1 |
| 400 | 51.5 | 83.1 | 83.2 | 13.9 | 2.9 | |
| 450 | 72.8 | 83.8 | 83.8 | 10.8 | 5.4 | |
| MoS2 nanoflake | 350 | 17.8 | 71.9 | 71.8 | 27.4 | 0.8 |
| 400 | 48.1 | 86.2 | 86.2 | 11.8 | 2.0 | |
| 450 | 66.3 | 87.6 | 87.5 | 8.3 | 4.2 | |
As shown in Table 2, under the same hydrocracking conditions, the order of conversion of DPM in the presence of different MoS2 products from high to low is MoS2 nanosphere, MoS2 nanoflake, MoS2 microsphere, which is consistent with the order of specific surface area of the MoS2 products. MoS2 with high specific surface area can provide more coordinatively unsaturated sites and yield more hydrogen free-radicals to participate in the reaction. The MoS2 nanosphere delivered a higher catalytic activity in DPM hydrocracking than a FeS2 catalyst33 and a MoS2 catalyst produced by water-soluble Mo precursor.34 Liu et al. used the ratio of gross products of hydrosaturation and DPM conversion to describe the “hydrogenation activity” of the catalyst.34 Therefore, the prepared MoS2 nanosphere presents higher hydrogenation activity than the MoS2 microsphere and MoS2 nanoflake. Moreover, the catalytic activity difference between the MoS2 nanosphere and nanoflake was enhanced by high temperature. Table 2 also shows that the conversion of DPM increased with increasing reaction temperature in the presence of the same catalyst. This indicates that high temperature facilitates formation of hydrogen free-radicals to participate in DPM conversion.
The selectivity results of DPM hydrocracking in Table 2 show that the percentages of scission products (benzene and toluene) greatly increased with increasing temperature from 350 °C to 400 °C, but hardly increased with increasing temperature from 400 °C to 450 °C. However, the percentage of DHM continued to increase with the increasing temperature. These results reveal that scission of the Car–Calk bond dominates the reaction at high temperature, meanwhile the hydrogenation saturation abilities of MoS2 products were enhanced by high temperature. Additionally, at the same temperature, DPM conversion with MoS2 nanospheres showed higher yields of BCH and DHM, which indicates that the hydrogenation saturation ability of MoS2 nanospheres was higher than that of MoS2 nanoflakes and MoS2 microspheres.
Conclusions
In summary, MoS2 nanospheres with an average diameter of 400 nm and specific surface area of 67.9 m2 g−1 were successfully prepared via a solvothermal process assisted by [EMIM]Br in a mixed solvent of DMF–water (V1/V2 = 3/6) at 200 °C for 24 h. The ratio of DMF to water has a significant effect on the size and morphology of MoS2 products. The strategy described here could be extended to other transition metal sulfide nanomaterials. Additionally, the MoS2 nanospheres delivered high catalytic activity in the hydrocracking of diphenylmethane. The scission of the Car–Calk bond dominates the DPM conversion at high temperature. Meanwhile, the hydrogenation saturation abilities of MoS2 products were enhanced by high temperature, with that of MoS2 nanospheres being higher than those of MoS2 nanoflakes and MoS2 microspheres at the same temperature.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21176259), Shandong Provincial Natural Science Foundation, China (ZR2015BM003) and the Fundamental Research Funds for the Central Universities (15CX05009A).Notes and references
- J. Chen, S. Li, Q. Xu and K. Tanaka, Chem. Commun., 2002, 16, 1722–1723 RSC.
- L. Rapoport, Y. Bilik, Y. Feldman, M. Homyonfer, S. R. Cohen and R. Tenne, Nature, 1997, 387, 791–793 CrossRef CAS.
- Q. Wang and J. Li, J. Phys. Chem. C, 2007, 111, 1675–1682 CAS.
- J. Chen, N. Kuriyama, H. Yuan, H. T. Takeshita and T. Sakai, J. Am. Chem. Soc., 2001, 123, 11813–11814 CrossRef CAS.
- S. J. Tauster, T. A. Percoraro and R. R. Chianelli, J. Catal., 1980, 63, 515–519 CrossRef CAS.
- R. R. Chianelli, A. F. Ruppert, S. K. Behal, B. H. Kear, A. Wold and R. Kershaw, J. Catal., 1985, 92, 56–63 CrossRef CAS.
- C. T. Tye and K. J. Smith, Catal. Lett., 2004, 95, 203–209 CrossRef CAS.
- C. T. Tye and K. J. Smith, Catal. Today, 2006, 116, 461–468 CrossRef CAS PubMed.
- P. M. Mortensen, J.-D. Grunwaldt, P. A. Jensen, K. G. Knudsen and A. D. Jensen, Appl. Catal., A, 2011, 407, 1–19 CrossRef CAS PubMed.
- V. P. Santos, B. van der Linden, A. Chojecki, G. Budroni, S. Corthals, H. Shibata, G. R. Meima, F. Kapteijn, M. Makkee and J. Gascon, ACS Catal., 2013, 3, 1634–1637 CrossRef CAS.
- M. Soto-Puente, M. Del Valle, E. Flores-Aquino, M. Avalos-Borja, S. Fuentes and J. Cruz-Reyes, Catal. Lett., 2007, 113, 170–175 CrossRef CAS.
- G. Bellussi, G. Rispoli, D. Molinari, A. Landoni, P. Pollesel, N. Panariti and E. Montanari, Catal. Sci. Technol., 2013, 3, 176–182 CAS.
- G. Bellussi, G. Rispoli, A. Landoni, R. Millini, D. Molinari, E. Montanari and P. Pollesel, J. Catal., 2013, 308, 189–200 CrossRef CAS PubMed.
- R. R. Chianelli, M. H. Siadati, M. P. de la Rosa, G. Berhault, J. P. Wilcoxon, R. Bearden Jr and B. L. Abrams, Catal. Rev., 2006, 48, 1–41 CAS.
- D. Wang, Z. Pan, Z. Wu, Z. Wang and Z. Liu, J. Power Sources, 2014, 264, 229–234 CrossRef CAS PubMed.
- L. Ma, W.-X. Chen, H. Li, Y.-F. Zheng and Z.-D. Xu, Mater. Lett., 2008, 62, 797–799 CrossRef CAS PubMed.
- L. Ma, L. M. Xu, X. P. Zhou and X. Y. Xu, Mater. Lett., 2014, 132, 291–294 CrossRef CAS PubMed.
- D. Mahajan, C. L. Marshall, N. Castagnola and J. C. Hanson, Appl. Catal., A, 2004, 258, 83–91 CrossRef CAS PubMed.
- N. Savjani, E. A. Lewis, R. A. D. Pattrick, S. J. Haigh and P. O'Brien, RSC Adv., 2014, 4, 35609–35613 RSC.
- Y. H. Lee, X. Q. Zhang, W. Zhang, M. T. Chang, C. T. Lin, K. D. Chang, Y. C. Yu, J. T. W. Wang, C. S. Chang, L. J. Li and T. W. Lin, Adv. Mater., 2012, 24, 2320–2325 CrossRef CAS PubMed.
- J. Wang, L. Chen, W. Lu, M. Zeng, L. Tan, F. Ren, C. Jiang and L. Fu, RSC Adv., 2015, 5, 4364–4367 RSC.
- H. Liao, Y. Wang, S. Zhang and Y. Qian, Chem. Mater., 2001, 13, 6–8 CrossRef CAS.
- D. Vollath and D. V. Szabó, Mater. Lett., 1998, 35, 236–244 CrossRef CAS.
- M. Remškar, A. Mrzel, M. Viršek and A. Jesih, Adv. Mater., 2007, 19, 4276–4278 CrossRef PubMed.
- M. Rajamathi and R. Seshadri, Curr. Opin. Solid State Mater. Sci., 2002, 6, 337–345 CrossRef CAS.
- T. Wang, H. Kaper, M. Antonietti and B. Smarsly, Langmuir, 2007, 23, 1489–1495 CrossRef CAS PubMed.
- T. Zhang, H. Guo and Y. M. Qiao, J. Lumin., 2009, 129, 861–866 CrossRef CAS PubMed.
- H. Luo, C. Xu, D. Zou, L. Wang and T. Ying, Mater. Lett., 2008, 62, 3558–3560 CrossRef CAS PubMed.
- N. Li, Y. Chai, Y. Li, Z. Tang, B. Dong, Y. Liu and C. Liu, Mater. Lett., 2012, 66, 236–238 CrossRef CAS PubMed.
- H. Li, W. Li, L. Ma, W. Chen and J. Wang, J. Alloys Compd., 2009, 471, 442–447 CrossRef CAS PubMed.
- Y. Shen, H. Hoffmann and J. Hao, Langmuir, 2009, 25, 10540–10547 CrossRef CAS PubMed.
- E. Furimsky, Catalysts for upgrading heavy petroleum feeds, Elsevier, 2007 Search PubMed.
- X. Y. Wei, E. Ogata, Z. M. Zong and E. Niki, Energy Fuels, 1992, 6, 868–869 CrossRef CAS.
- D. Liu, X. Kong, M. Li and G. Que, Energy Fuels, 2009, 23, 958–961 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures. See DOI: 10.1039/c5ra08424e |
| This journal is © The Royal Society of Chemistry 2015 |