
Hybrid structures of a BN nanoribbon/single-walled carbon nanotube: ab initio study†
Abstract
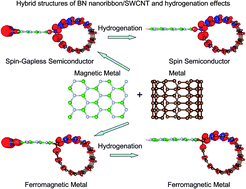
Hybrid structures of a zigzag edge BN nanoribbon/single-walled carbon nanotube, namely, (i) ZBNNR-B-SWCNT in which only the B-edge of a zigzag edge BN nanoribbon (ZBNNR) is passivated with a single-walled carbon nanotube (SWCNT); (ii) ZBNNR-N-SWCNTs in which only the N-edge of ZBNNR is terminated with a SWCNT; (iii) hydrogenated ZBNNR-B-SWCNT; and (iv) hydrogenated ZBNNR-N-SWCNT, have been studied via standard spin-polarized density functional theory (DFT) calculations as well as ab initio molecular dynamics (MD) simulations. The DFT calculations and ab initio MD simulations results clearly show that all the examined hybrid structures are stable at room temperature. The formation energy, as well as the local DOS and Mulliken charge analysis, reveals that the hybrid structure of ZBNNR-B-SWCNT is more stable than that of ZBNNR-N-SWCNT, since the covalent bond of B–C is stronger than that of N–C owing to the electronegativity difference of the N and C atoms (1.49) being larger than that of the C and B atoms (0.51). Surprisingly, the ZBNNR-B-SWCNTs belong to intrinsic ferromagnetic metals, whereas the ZBNNR-N-SWCNTs belong to intrinsic spin-gapless semiconductors (SGS). Furthermore, in contrast to hydrogenated ZBNNRs, which are nonmagnetic semiconductors, hydrogenated ZBNNR-N-SWCNTs turn into intrinsic spin-semiconductors (hydrogenation induces a SGS–spin-semiconductor transition), whereas hydrogenated ZBNNR-B-SWCNTs remain in the ferromagnetic metallic state, because H-passivation removes the dangling bond states of N-edge or B-edge, but the magnetic properties of the “-SWCNT” edge remain unchanged.
 Please wait while we load your content...
Please wait while we load your content...

