
Influence of a blend of guar gum and poly(vinyl alcohol) on long term stability, and antibacterial and antioxidant efficacies of silver nanoparticles†
Abstract
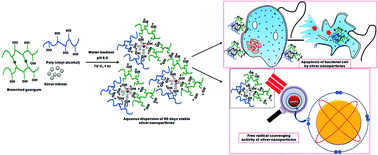
This paper describes a detailed procedure for the synthesis of highly stable (almost for 60 days) and monodispersed silver nanoparticles (average particle size of 16 nm which changed to 42 nm only on the 60th day of standing under refrigeration) in the presence of a blend of a polysaccharide (guar gum) and a hydrophilic synthetic polymer (poly(vinyl alcohol)) where both the polymers provided synergistic effects in reduction and stabilization of incipient nanoparticles. Time dependant stability of aqueous dispersions of silver nanoparticles in various compositions of polymeric matrices was studied from UV-Vis spectroscopy, DLS and zeta potential characterization data. An FTIR spectrum and HRTEM image along with EDX results of the best optimized compositions in respect of particle size and particle size distribution of silver nanoparticles were also obtained. A study displayed how viscosity of the polymeric matrices played a vital role in the long term stabilization of aqueous dispersions of silver nanoparticles. For the first time we are reporting that silver nanoparticles synthesized by a green technique, using a blend of two highly viscous polymers, exhibited excellent stability. The aqueous dispersions of silver nanoparticles also displayed strong antibacterial and antioxidant properties. Hence our synthesized silver nanoparticles may be suitable for commercialization in future.
 Please wait while we load your content...
Please wait while we load your content...

