
Graphene oxide wrapped individual silver nanocomposites with improved stability for surface-enhanced Raman scattering†
Nan Gao,
Ting Yang,
Tao Liu,
Yu Zou and
Jiang Jiang*
CAS Key Laboratory of Nano-Bio Interface, i-Lab and Division of Nanobiomedicine, Suzhou Institute of Nano-Tech and Nano-Bionics, Chinese Academy of Sciences, Suzhou 215123, China. E-mail: jjiang2010@sinano.ac.cn
First published on 18th June 2015
Abstract
As one of the best materials for surface-enhanced Raman scattering (SERS), Ag suffers from its tendency to oxidation, posing serious limitations for its use as a reliable long-term SERS substrate. Graphene oxide (GO) is considered to be a promising SERS-active platform due to the observed chemical enhancement originating from the interaction between probe molecules and oxygen containing functional groups on its surface. Herein, we present the synthesis of core–shell GO wrapped individual Ag nanocomposites (NCs) by electrostatic assembly of GO on SiO2@Ag nanostructures. The SERS enhancement factor (EF) of probe molecules on SiO2@Ag@GO is 1.8 times that on SiO2@Ag, due to the chemical enhancement brought upon by GO. Moreover, the GO surrounding the SiO2@Ag nanoparticles (NPs) shields Ag from oxidation, making them remain stable and display highly retained SERS activities even after long-term storage, while the bare SiO2@Ag NPs would have lost ∼80% of the original activity after the same treatment. As the NCs have displayed enhanced and stable SERS activities, colloidal encapsulation by GO has been proven to be an efficient way to prepare a SERS substrate with long-term stability for practical applications.
Introduction
Raman spectroscopy has incomparable advantages in characterizing material structures due to its precise molecular identification with multiplexing capabilities,1 and the discovery of surface-enhanced Raman scattering (SERS) phenomena2,3 has overcome the limitation of the intrinsic low Raman scattering cross-section, opening the doors for its applications in trace analyte detection.4–6Other than the first reported roughened Ag electrode, SERS substrates have been obtained in many different ways, including but not limited to, electrochemical deposition,7 chemical vapor deposition (CVD),8 micro and nano fabrication,9 and synthesis of colloidal nanostructures.10 Noble metal (Ag and Au) colloidal nanocrystals of various morphologies (nanoparticles, nanocubes, nanoplates, and nanostars) have been widely used as SERS substrates, due to their ease of preparation and ability to support localized surface plasmon resonances in the visible spectrum.11 Compared to Au, whose plasmonic resonance is damped by the interband transitions, Ag is thought to be an ideal Raman enhancing material with high efficacy.12 However, Ag suffers from poor stability, as it can be easily oxidized under ambient conditions, leading to reduced intensity and poor reproducibility of Raman signals, posing a serious limitation for its reliable long-term usage in SERS applications.
Recently, graphene has been demonstrated as an ultrathin anti-corrosion coating for metals that are prone to oxidations.13 As a derivative of graphene, graphene oxide (GO) has abundant oxygen-containing functional groups (such as epoxy, hydroxyl and carboxyl groups) existing on its surface, improving its water solubility and enabling its usage in many biological applications such as bio-sensing, bio-imaging, drug delivery, and photothermal therapy.14–17 Moreover, graphene and GO are also promising SERS substrate due to the observed chemical enhancement and molecular enriching effect arising from strong π–π interactions.18–21
Due to the above mentioned reasons, studies on nanocomposites (NCs) composed of Ag nanoparticles (NPs) and graphene or its derivatives have gained a lot of attention in the past few years.22–43 For example, Ag decorated GO or GO covered Ag substrates have been used for detecting aromatic molecules, taking advantage of the strong interaction between these molecules with graphene surface.32–35 Graphene and GO have also acted as ultrathin spacers for the construction of SERS hot spots with good reproducibility.36,37 Very recently, CVD grown graphene on top of noble metal nanostructures as a pinhole-free isolating shell has been demonstrated as an effective method to improve stability and enhance sensitivity of the SERS substrate.38–40,42 In addition, sandwich structures have also been constructed where Ag nanoparticles were placed in between GO sheaths.41
Previous studies have focused mainly on coating Ag NPs dispersed on a planar surface, and work on individually wrapped Ag nanostructures has been rare. Compared with planar structure of Ag–GO nanocomposites, core–shell structure of Ag@GO nanocomposites can be more versatile SERS substrates, with colloidal dispersion stability and possibility to include magnetic cores for additional magnetic enrichment functions.
Herein, we present work on constructing colloidal core–shell GO wrapped Ag nanostructures by coating a thin layer of SERS-active material GO outside Ag NPs. By first synthesizing SiO2@Ag nanostructures and modifying the surface with a polyelectrolyte to make them positively charged, negatively charged GO can then be assembled quickly onto this oppositely charged SiO2@Ag surface. Raman probe molecules on these core–shell SiO2@Ag@GO NCs have exhibited stronger Raman signals than that on SiO2@Ag. The stability of the as-prepared SERS substrates was then investigated by exposing them to ambient environment for 50 days.
Experimental section
Materials
Tetraethyl orthosilicate (TEOS, Aladdin), (3-aminopropyl) triethoxysilane (APTES, Aladdin), silver nitrate (AgNO3, AR, Aladdin), polyvinylpyrrolidone (PVP, K30, Sinopharm Chemical Reagent Co., Ltd), potassium borohydride (KBH4, 95%, Sinopharm Chemical Reagent Co., Ltd), poly(diallyldimethylammonium chloride) (PDDA, Mw < 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Aladdin), formaldehyde solution (HCHO, AR, Sinopharm Chemical Reagent Co., Ltd), ammonia solution (NH3·H2O, AR, Sinopharm Chemical Reagent Co., Ltd), ethanol (C2H5OH, AR, Sinopharm Chemical Reagent Co., Ltd), and 4-aminothiophenol (4-ATP, Aladdin) were all used without any further purification. Deionized water was used in all cases.
000, Aladdin), formaldehyde solution (HCHO, AR, Sinopharm Chemical Reagent Co., Ltd), ammonia solution (NH3·H2O, AR, Sinopharm Chemical Reagent Co., Ltd), ethanol (C2H5OH, AR, Sinopharm Chemical Reagent Co., Ltd), and 4-aminothiophenol (4-ATP, Aladdin) were all used without any further purification. Deionized water was used in all cases.
Characterization
UV-vis absorption spectra were recorded on a Lambda-25 spectrometer (PerkinElmer, USA), and zeta potentials were recorded on Malvern ZetaSizer Nano ZS90. Transmission electron microscopy (TEM) characterization was performed on a Tecnai G2 F20 S-Twin TEM (FEI, USA) operating at 200 kV. TEM samples were prepared by drying a drop of solution in a dark room on amorphous carbon-coated copper grids. Scanning electron microscopy (SEM) characterization was performed on QUANTA 400 (FEI, USA). Raman spectra were recorded on a Jobin-Yvon LabRam HR 800 confocal micro-Raman system with the excitation wavelength of 532 nm, and all the Raman spectra were collected with 5 s integration time, unless otherwise noted.Synthesis of GO nanosheets
Synthesis of graphene oxide nanosheets (NSs) from natural graphite flakes was conducted according to the procedures reported in our previous work,44 adopting a modified Hummers method originally presented by Kovtyukhova et al.45 The as-prepared GO can be stably dispersed in water for several months.Synthesis of SiO2@Ag NPs
SiO2@Ag NPs were synthesized via a seed-mediated growth approach. SiO2 NPs were prepared by the Stöber method, where 5 mL TEOS was added to the mixture of ethanol (75 mL), deionized water (10 mL) and ammonia solution (10 mL) with quick stirring for 7 h. The as formed SiO2 NPs were washed by excess ethanol to remove unreacted TEOS, and finally dried at 40 °C in a drying oven.The primary and key procedure for coating SiO2 NPs with Ag is the synthesis of uniform silver seeds with high density on SiO2 NPs. Firstly, 20 mg SiO2 NPs were dispersed in 2 mL ethanol, which was then added to 2 mL PVP solution (5 mg mL−1). After 15 min sonication, the mixture was washed with deionized water by centrifugation for 3 times and sonicated in 4 mL ethanol. Subsequently, 1 mL Tollens' reagent ([Ag(NH3)2]OH, 0.02 mmol mL−1) was added to SiO2 NPs suspension with continuous stirring for 15 min. Excess [Ag(NH3)2]+ was removed by centrifugation for 3 times. The obtained NPs were redispersed in 30 mL ethanol in an ice bath, and then 1 mL KBH4 (0.5 mg mL−1) was quickly injected into the solution. After nearly 15 min, the solution turned from light yellow to dark brown. SiO2@Ag seeds could be gathered by further centrifugation for 10 min at 5000 rpm. This procedure was repeated 3 times in order to remove free Ag NPs in solution.
In the following Ag seeds growth process, half of the as-prepared SiO2@Ag seeds were added to the 200 mL 0.25% PVP solution with 10 mg AgNO3. Then 0.3 mL formaldehyde and 0.6 mL ammonia solution were quickly injected in sequence. The color of solution changed to bluish grey in a few seconds. The reaction was allowed to proceed for 1 h, and then SiO2@Ag NPs can be obtained after 3 times centrifugation at 4000 rpm. Finally, SiO2@Ag NPs were dispersed in 10 mL ethanol.
Fabrication of SiO2@Ag@GO NCs
PDDA was used for coating the negatively charged SiO2@Ag NPs to reverse their surface potential. 1 mL PDDA (1 mg mL−1) was mixed with 1 mL SiO2@Ag NPs under magnetic stirring for 15 min. The solution was then centrifuged and redispersed in 4 mL ethanol. 1 mL GO (0.2 mg mL−1) was added to this positively charged SiO2@Ag NPs solution, and sonicated for 5 min. SiO2@Ag@GO NCs were obtained as precipitates after several rounds of centrifugations.Preparation of SERS substrates
Si wafers were first cut into small pieces of 1 cm × 1 cm before acid treatment, which were then washed in acetone, ethanol and water, and further cleaned in Piranha solution (H2SO4:H2O2 = 3:1) at mild stirring conditions for 2 h. After washing by water and ethanol, the Si substrates were immersed into ethanol solution of APTES, and heated at 90 °C under refluxing for 24 h. APTES modified Si substrates were washed with ethanol and water again before use. These APTES modified Si substrates and original Si substrates were immersed into 1 mL SiO2@Ag NPs and SiO2@Ag@GO NCs respectively for 2 h. Then, SERS substrates were washed with ethanol to remove unabsorbed NPs and NCs. The SiO2@Ag NPs and SiO2@Ag@GO NCs SERS substrates were then immersed into 1 mL 4-ATP solution at four different concentrations (10−4 M, 10−5 M, 10−6 M and 10−7 M) for 12 h. Finally, the as-prepared SERS substrates were dried in air for Raman detection.
The SERS activities of SiO2@Ag NPs and SiO2@Ag@GO NCs substrates were collected using a 532 nm laser at 6.5 mW, with a 50× objective. The integration time was 5 s.
Results and discussion
Synthesis of SiO2@Ag@GO nanocomposites
The five steps (I–V) illustrated in Fig. 1 depict the formation of SiO2@Ag@GO NCs, which are critical in synthesizing the nanocomposites with uniform morphology.46,47 In step I, 500 nm monodisperse SiO2 NPs with narrow size distributions were first synthesized, and then served as templates to grow Ag seed NPs. Considering the size of GO served as the protecting layer outside Ag NPs, uniform Ag NPs of several hundred nanometers in size are difficult to obtain without the SiO2 template. Moreover, SiO2 templated growth will render Ag nanoparticle shells with strong scattering capability and broad scattering spectrum. Diameter of SiO2 NPs can be adjusted by varying reaction time, temperature, pH, and water/ethanol ratios. It was reported that Ag NPs with sizes ranged from 30 nm to 70 nm in Ag aggregates have the highest Raman enhancement factor (EF), and a plenty of “hot spots” could be created to enhance the SERS activity.48 However, since larger Ag NPs with uniform morphology and narrow size distribution were difficult to be synthesized in a single step, we adopted seed-mediated growth method to grow Ag on SiO2 NPs.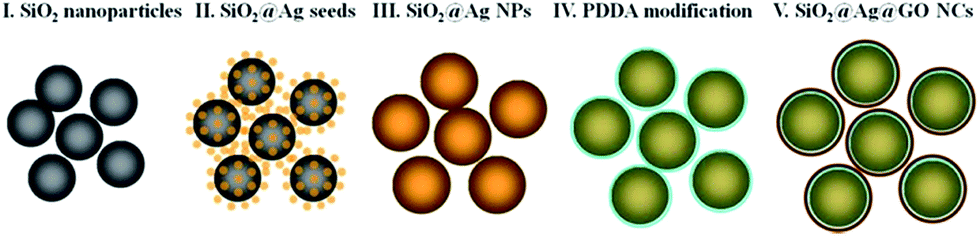 | ||
| Fig. 1 Schematic illustration of SiO2@Ag@GO NCs formation. | ||
In step II, Ag was reduced by KBH4 and grew to 5 nm Ag seed NPs on SiO2. As shown in Fig. 2A, small Ag seeds were formed on the surface of SiO2 NPs with high density. PVP, which has strong affinity to Ag, must be added to SiO2 colloids as a stabilizer before the nucleation of Ag NPs. Otherwise, a mass of Ag NPs would fall off from the surfaces of SiO2 NPs and began to agglomerate and grow to larger NPs (ESI Fig. S1†). Free Ag NPs in solution must be removed by centrifuging several times before being used in the following steps.
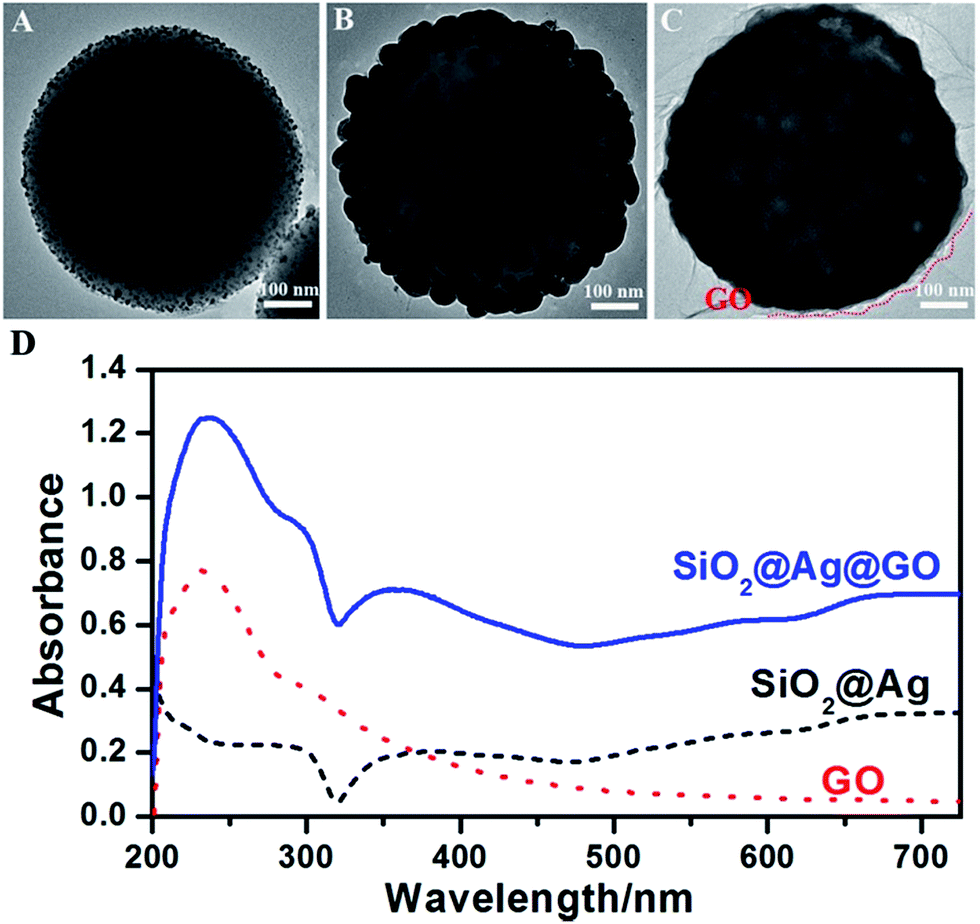 | ||
| Fig. 2 TEM images of (A) SiO2@Ag seeds, (B) SiO2@Ag NPs, and (C) SiO2@Ag@GO NCs; (D) the UV-vis absorption spectra of SiO2@Ag, GO, and SiO2@Ag@GO. | ||
50 nm Ag NPs surrounding SiO2 NPs were then synthesized via Ag seeded growth in step III, and Fig. 2B shows the TEM image of SiO2@Ag NPs. Ag+ was reduced by formaldehyde in a few seconds after the addition of ammonia solution, and the color of solution changed to bluish grey quickly. The reaction was continued to react for 1 h, and ultimately dense and uniform Ag NP with average size of 50 nm were obtained on SiO2 templates.
Electrostatic assembly of SiO2@Ag NPs and GO NSs was used to prepare SiO2@Ag@GO NCs (step IV). A cationic polyelectrolyte-PDDA was used for surface modification, since similarly negatively charged SiO2@Ag NPs and GO NSs were difficult to be self-assembled. After PDDA modification, SiO2@Ag NPs became positively charged, which made negatively charged GO being assembled easily forming a core–shell structure, and this process was monitored by zeta potential measurements shown in Fig. S2.† Due to their large sizes, SiO2@Ag NPs and SiO2@Ag@GO NCs will settle under gravitation eventually, which can be redispersed by sonication. Fig. 2C shows the morphology of GO wrapped Ag NCs under TEM, where the low contrast nanosheets marked by red dashed line indicated the existence of GO surrounding the SiO2@Ag NPs, and the corresponding UV-vis absorption spectra is shown in Fig. 2D. The absorption peak at 230 nm, coming from the π–π* transition of sp2 carbon, is the characteristic absorption peak of GO. The appearance of this characteristic peak after assembling SiO2@Ag NPs with GO confirmed the formation of SiO2@Ag@GO NCs. The characteristic absorption peak of Ag can be barely observed from SiO2@Ag NPs, likely due to the coupling between neighboring Ag nanoparticles inside the shell region, and large scattering from the SiO2 and Ag shell. Similar observation was also reported by Li and co-workers.47
Next, SERS substrates were fabricated by electrostatic absorption of SiO2@Ag NPs or SiO2@Ag@GO NCs on Si wafers (1 cm × 1 cm). Positively charged SiO2@Ag@GO NCs could be assembled easily on negatively charged Si wafers via electrostatic absorption, and this produced sub-monolayer SiO2@Ag@GO NCs with no obvious stacking as shown in Fig. 3A. Because of the close integration of GO and SiO2@Ag NPs, GO had almost a full coverage outside SiO2@Ag NPs, leading to difficulty in identifying low contrast GO under SEM. However, the existence of GO wrapped around the SiO2@Ag NPs can be seen more obviously in SEM image from the junctions between adjacent SiO2@Ag@GO NCs (Fig. 3B). Unlike positively charged SiO2@Ag@GO NCs, however, negatively charged SiO2@Ag NPs were difficult to load on Si substrates uniformly because of the repelling force between like charges. If the negatively charged Si substrates were immersed in SiO2@Ag solution, deposition of NPs by gravitation on Si substrates would lead to the stacking of NPs forming multilayered NPs aggregates. Thus, for SiO2@Ag NPs deposition, Si substrates were modified by positively charged APTES by refluxing in the ethanol solution of APTES for 24 h. Then, strong electrostatic attraction between NPs and Si substrates would facilitate the formation of SERS substrates with relatively uniform distribution (Fig. 3C and D).
 | ||
| Fig. 3 SEM images of (A and B) SiO2@Ag@GO NCs and (C and D) SiO2@Ag NPs absorbed on Si wafers. (B) and (D) are higher magnification SEM images of (A) and (C), respectively. | ||
SERS activity
To investigate the SERS activities of SiO2@Ag NPs and SiO2@Ag@GO NCs, 4-ATP of four different concentrations (10−4 M, 10−5 M, 10−6 M, and 10−7 M) were chosen as Raman probe for further analysis (the obtained SERS spectra are shown in Fig. 4). The peaks observed at 1076 cm−1, 1189 cm−1 and 1578 cm−1 were intrinsic Raman peaks of 4-ATP, with the Raman spectrum of pure 4-ATP on Si shown in Fig. S3† collected as a reference, in accordance with previous reports.49 The Raman peaks of D band (1355 cm−1) and G band (1580 cm−1) of GO could be well observed on SiO2@Ag@GO substrate (Fig. S4†). However, Raman spectrum of SiO2@Ag substrate also showed a relatively low intensity broad peak at around 1600 cm−1, likely coming from PVP, which has a characteristic Raman peak at 1606 cm−1 and 1640 cm−1.50 During the SERS measurement, a new Raman peak appeared at 1142 cm−1 was assigned to 4, 4′-dimercaptoazobenzene (DMAB), an oxidative coupling dimer of 4-ATP formed under intense laser irradiation.49,51,52 The fingerprint signal of DMAB at 1139 cm−1 can also be recognized even when the laser power was set as low as 0.01 mW. Taking into consideration that the broad Raman peaks of GO and PVP at around 1600 cm−1 could interfere with the identification and quantification of the Raman fingerprint peak of 4-ATP at 1578 cm−1, we evaluated the SERS activities of 4-ATP on SiO2@Ag and SiO2@Ag@GO substrates by comparing their relative Raman intensities at 1076 cm−1, a strong and characteristic Raman peak of 4-ATP, originating from a combination of phenyl breathing, C–H bending, and C–S stretching modes.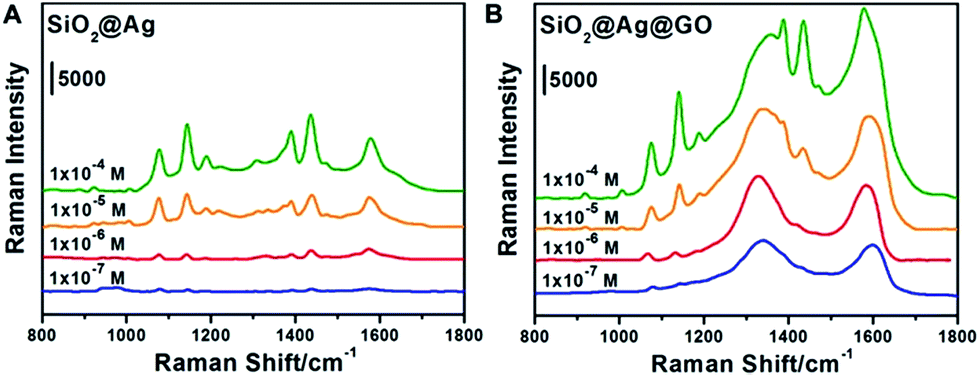 | ||
| Fig. 4 SERS spectra of different concentrations (10−4 M, 10−5 M, 10−6 M, and 10−7 M) of 4-ATP absorbed on (A) SiO2@Ag and (B) SiO2@Ag@GO substrates. All the spectra are plotted at the same scale, with vertical positions moved arbitrarily for better viewing purpose. | ||
Fig. 4 compares the relative SERS intensities of 4-ATP on SiO2@Ag and SiO2@Ag@GO substrates, demonstrating the effect of GO on the Raman activities of the probe molecules. As shown in Fig. 4A, strong Raman scattering at 1076 cm−1 was obtained when the concentration of 4-ATP was 10−4 M. As a comparison, we found that the relative Raman intensity of the same concentration 4-ATP on SiO2@Ag@GO substrate was about 1.4 fold higher than that of 4-ATP on SiO2@Ag substrate, showing enhanced Raman signals in the presence of the GO coating (Fig. 4B). The Raman intensities went down accordingly as the concentrations of 4-ATP were decreased, with only a weak scattering peak at 1076 cm−1 and broad peak of D and G bands of GO observed.
The SERS enhancement factor (EF) for 4-ATP on different substrates can be calculated according to the equation EF = ISERSNbulk/IbulkNsurface, where ISERS stands for the Raman intensities of 10−4 M 4-ATP on SERS substrates, and Ibulk represents the Raman intensity of 1 M 4-ATP on a Si substrate. In order to eliminate the effect of plasmon coupling between neighboring nanoparticles and quantify the EF of SiO2@Ag and SiO2@Ag@GO more accurately, we selected only single nanoparticles (diameter of laser spot is ∼1 μm, and SiO2@Ag, SiO2@Ag@GO were about 600 nm in size) for Raman detection. Ibulk at 1076 cm−1 was measured to be 12990 (Fig. S3†). ISERS of SiO2@Ag and SiO2@Ag@GO substrates were obtained by measuring the Raman signals from SiO2@Ag and SiO2@Ag@GO substrates after immersing them in 10−4 M 4-ATP solution for 12 h (Fig. S5,† ISERS-SiO2@Ag = 6513 and ISERS-SiO2@Ag@GO = 9242).
Nbulk and Nsurface were the number of 4-ATP molecules in the bulk sample within laser spot and the number of adsorbed 4-ATP molecules excited by laser, respectively. Nsurface can be determined by monitoring the UV-vis absorption spectra of free 4-ATP in solutions. UV-vis absorption spectra of 4-ATP at different concentrations ranging from 5 μM to 100 μM were first measured (Fig. S6A†), which were then used to obtain a calibration curve using the peak absorption at 257 nm (Fig. S6B†). After incubating SiO2@Ag and SiO2@Ag@GO with 10−4 M 4-ATP solutions for 12 h, the unabsorbed 4-ATP molecules were isolated in the supernatant by centrifugation (Fig. S5A†), from which the concentration of free 4-ATP was determined using the calibration curve. The number of 4-ATP molecules adsorbed on surface of SiO2@Ag and SiO2@Ag@GO were then calculated to be 1.07 × 1016 and 8.58 × 1015, respectively. More 4-ATP molecules were absorbed on SiO2@Ag surface, likely due to the strong binding between Ag surface and thiol molecules, compared to the π–π stacking force between 4-ATP and GO, favoring the absorption of 4-ATP on SiO2@Ag than those on SiO2@Ag@GO. Taking into account of the diameter (500 nm) and density (2.2 mg cm−3) of SiO2, the number of SiO2 templates was calculated to be 4.02 × 1012. Thus, on average, each single SiO2@Ag nanoparticle absorbed 2.667 × 103 4-ATP molecules (Nsurface-SiO2@Ag) and SiO2@Ag@GO absorbed 2.134 × 103 4-ATP molecules on its surface (Nsurface-SiO2@Ag@GO). Taking the laser spot size (1 μm in diameter), and the concentration of 4-ATP (1 M, 10 μL) into account, Nbulk had a value of 9.37 × 108. The EF was then calculated to be 7.81 × 105 for SiO2@Ag@GO, which was 1.8 fold higher than that of SiO2@Ag (EFSiO2@Ag = 4.40 × 105). As Nsurface-SiO2@Ag@GO is actually lower, it is the chemical enhancement of GO, rather than molecular enrichment effect, which made the Raman signals on SiO2@Ag@GO stronger in this case. Also, we carried on a control experiment by detecting the SERS activity of 10−4 M 4-ATP on SiO2@GO substrate. Fig. S7† showed no obvious Raman peak of 4-ATP under different concentration of 4-ATP, indicating the low EF of plain GO. SiO2@Ag@GO also showed good SERS performance toward other frequently used Raman probes, such as aromatic crystal violet (CV) and rhodamine B (RhB) molecules (Fig. S8†).
The thickness of GO coated on the surface of SiO2@Ag is hard to control because of the difficulties in quantifying GO nanosheets in the solution, as well as the follow-up characterization of its thickness. However, after a series of experiments and comparison, we choose GO at 0.2 mg mL−1 as an ideal reaction concentration. The SiO2@Ag@GO NCs substrates performed the best SERS activity at the concentration of 0.2 mg mL−1 compared with SiO2@Ag@GO NCs at the lower GO concentration (Fig. S9†). Concentration of GO higher than 0.2 mg mL−1 could decrease the SERS signal of 4-ATP absorbing on the surface of SiO2@Ag@GO NCs, and magnify the SERS signal of GO because of the increasing thickness of GO coated on SiO2@Ag NPs. In order to show the reproducibility of the as-made SERS substrates, we chose 10 random spots from different batches of SERS substrate samples, and their SERS activities are shown in Fig. S10.† From the comparison of SERS activities of SiO2@Ag and SiO2@Ag@GO (Fig. S10C†), SiO2@Ag@GO NCs showed slightly higher reproducibility compared with SiO2@Ag NPs, and their difference is statistically significant (p < 0.05).
Long-term SERS activity
To investigate the stability and the effect of possible protection of Ag against oxidation with a thin layer of GO coating, SERS performance of SiO2@Ag@GO substrates were evaluated after being stored under ambient condition for a period of time. For comparison, the starting SiO2@Ag NPs were chosen as the control group. As shown in Fig. 5, Raman intensities of probe molecules decreased continuously as time went by on both substrates due to Ag oxidation. After 8 days of storage, ∼85% original Raman signals remained for SiO2@Ag@GO substrate, while less than 70% Raman signals were detected on SiO2@Ag (Fig. 5C). The Raman signals went down even further with prolonged storage period. At the end of 50 days under ambient condition, Raman peak intensities of ∼50% of the original value can still be observed on SiO2@Ag@GO substrate, while less than 20% of the original signals were left for SiO2@Ag substrate, demonstrating the relatively lower stability of SiO2@Ag NPs compared to SiO2@Ag@GO NCs. We calculated the long-term enhancement factor (L-EF) of SERS substrates after 50 days of exposure and found that L-EF of SiO2@Ag@GO NCs (L-EFSiO2@Ag@GO = 3.95 × 105) was about 5.3 times higher than L-EF of SiO2@Ag NPs (L-EFSiO2@Ag = 7.45 × 104). Our experimental results demonstrated that SiO2@Ag@GO NCs with higher stability due to GO protection against oxidation could serve as a good candidate for long-term SERS substrate. A full coverage of GO outside SiO2@Ag can prevent the direct exposure of SiO2@Ag NPs to ambient external environment (oxygen and water), thus slowing down the oxidation and degradation of Ag. The observed loss of SERS performance on SiO2@Ag@GO NCs might be due to incomplete coverage of GO, and domain boundary between GO NSs. Furthermore, oxygen containing functional groups on GO surface could remove the reactive oxygen intermediates from the environment which can otherwise oxidize Ag.53 However, the intrinsic broad Raman scattering of GO at ∼1600 cm−1 will limit SERS detection of analytes with only fingerprint peaks overlapping with this.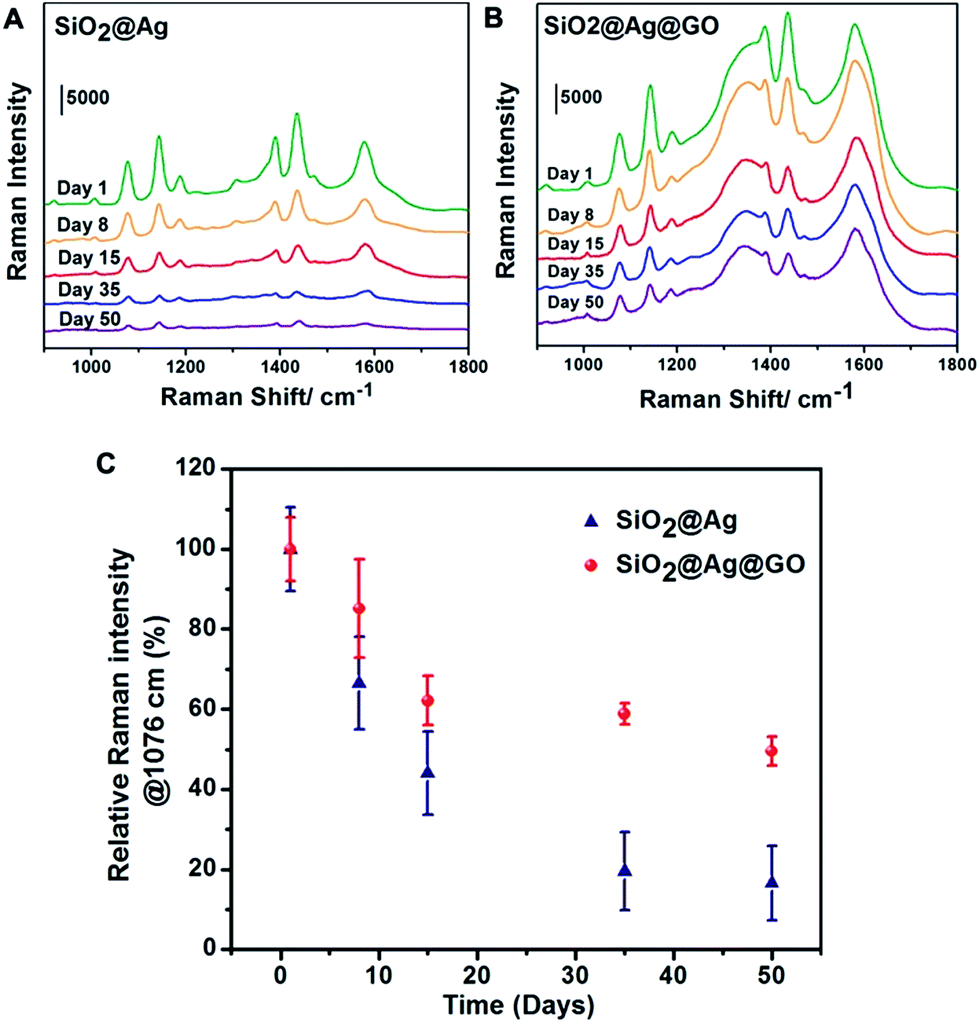 | ||
| Fig. 5 SERS spectra of 4-ATP on (A) SiO2@Ag and (B) SiO2@Ag@GO substrates, which were exposed to ambient condition for 1, 8, 15, 35 and 50 days. All the spectra are plotted at the same scale, with vertical positions moved arbitrarily for better viewing purpose. (C) Plots of relative Raman intensities of 4-ATP at 1076 cm−1 on SiO2@Ag and SiO2@Ag@GO substrates, after they were exposed to ambient environment for different days. | ||
For the EF calculation, the effect of 4-ATP converting to DMAB was not accounted for, which could set the presented EF to be lower than the actual value. Moreover, the relative EF comparison between SiO2@Ag NPs and SiO2@Ag@GO NCs is valid, as the ratio between the characteristic Raman scattering peak of 4-ATP and DMAB was similar for both substrates (Fig. 4) at similar surface coverage. Our obtained single particle EF value is very close to what has been reported by Fan et al. on GO capped Ag octahedron NPs, and the extra enhancement factor brought by GO also agrees well with their report.28 With our SiO2@Ag@GO NCs left in ambient conditions for 50 days, the associated EF value is still close to the pristine SiO2@Ag NCs, demonstrating their potential use as highly sensitive stable SERS substrates. Unlike CVD based graphene coating, which can be made crack and hole free with additional deoxidation effect,42 colloidal preparation of GO encapsulation is easier to conduct, albeit with less ideal long-term stability; and it is possible to introduce more functionality, such as using Fe3O4@ Ag as the core for magnetic field enriched SERS detection schemes.54
Conclusions
We have successfully prepared core–shell SiO2@Ag@GO NCs by electrostatic assembly, and investigated their SERS enhancement factor and long-term stabilities. The additional chemical enhancement brought upon by GO has led to the increment of Raman enhancement factor on SiO2@Ag@GO by 1.8 fold compared to that on SiO2@Ag. More importantly, long-term Raman detection has proven that SiO2@Ag@GO is a more stable SERS substrate because GO can partially shield Ag from oxidation. After 50 days of exposure to the ambient condition, strong Raman scattering signals can still be observed on SiO2@Ag@GO substrate, which is not the case for bare SiO2@Ag; Raman signal of molecules on SiO2@Ag@GO has retained ∼50% of the original value, while that on SiO2@Ag has only less than 20% remained. Our results show GO not only serves as an additional SERS enhancement surface, but also a protecting sheath for Ag nanostructures, whereby stability of Ag under GO coating has improved greatly compared to that of the bare Ag surface.Acknowledgements
This work is funded by the “Hundred Talents” program of Chinese Academy of Sciences, and Natural Science Foundation of China (grant no. 21175148).Notes and references
- K. Kneipp, H. Kneipp, I. Itzkan, R. R. Dasari and M. S. Feld, Chem. Rev., 1999, 99, 2957–2976 CrossRef CAS PubMed.
- M. Fleischmann, P. J. Hendra and A. J. McQuillan, Chem. Phys. Lett., 1974, 26, 163–166 CrossRef CAS.
- D. L. Jeanmaire and R. P. Van Duyne, J. Electroanal. Chem. Interfacial Electrochem., 1977, 84, 1–20 CrossRef CAS.
- K. Kneipp, Y. Wang, H. Kneipp, L. T. Perelman, I. Itzkan, R. R. Dasari and M. S. Feld, Phys. Rev. Lett., 1997, 78, 1667–1670 CrossRef CAS.
- J. N. Anker, W. P. Hall, O. Lyandres, N. C. Shah, J. Zhao and R. P. Van Duyne, Nat. Mater., 2008, 7, 442–453 CrossRef CAS PubMed.
- X. M. Qian and S. M. Nie, Chem. Soc. Rev., 2008, 37, 912–920 RSC.
- P. He, H. Liu, Z. Li, Y. Liu, X. Xu and J. Li, Langmuir, 2004, 20, 10260–10267 CrossRef CAS PubMed.
- Z. Yuan, N. H. Dryden, J. J. Vittal and R. J. Puddephatt, Chem. Mater., 1995, 7, 1696–1702 CrossRef CAS.
- J. C. Hulteen, D. A. Treichel, M. T. Smith, M. L. Duval, T. R. Jensen and R. P. Van Duyne, J. Phys. Chem. B, 1999, 103, 3854–3863 CrossRef CAS.
- N. Leopold and B. Lendl, J. Phys. Chem. B, 2003, 107, 5723–5727 CrossRef CAS.
- Y. Wang, B. Yan and L. Chen, Chem. Rev., 2012, 113, 1391–1428 CrossRef PubMed.
- S. Abalde-Cela, P. Aldeanueva-Potel, C. Mateo-Mateo, L. Rodríguez-Lorenzo, R. A. Alvarez-Puebla and L. M. Liz-Marzán, J. R. Soc., Interface, 2010, 7, S435–S450 CrossRef CAS PubMed.
- S. Chen, L. Brown, M. Levendorf, W. Cai, S.-Y. Ju, J. Edgeworth, X. Li, C. W. Magnuson, A. Velamakanni, R. D. Piner, J. Kang, J. Park and R. S. Ruoff, ACS Nano, 2011, 5, 1321–1327 CrossRef CAS PubMed.
- M. Zhou, Y. Zhai and S. Dong, Anal. Chem., 2009, 81, 5603–5613 CrossRef CAS PubMed.
- X. Sun, Z. Liu, K. Welsher, J. Robinson, A. Goodwin, S. Zaric and H. Dai, Nano Res., 2008, 1, 203–212 CrossRef CAS PubMed.
- J. T. Robinson, S. M. Tabakman, Y. Liang, H. Wang, H. Sanchez Casalongue, D. Vinh and H. Dai, J. Am. Chem. Soc., 2011, 133, 6825–6831 CrossRef CAS PubMed.
- L. Zhang, J. Xia, Q. Zhao, L. Liu and Z. Zhang, Small, 2010, 6, 537–544 CrossRef CAS PubMed.
- W. Xu, X. Ling, J. Xiao, M. S. Dresselhaus, J. Kong, H. Xu, Z. Liu and J. Zhang, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 9281–9286 CrossRef CAS PubMed.
- W. Xu, N. Mao and J. Zhang, Small, 2013, 9, 1206–1224 CrossRef CAS PubMed.
- X. Yu, H. Cai, W. Zhang, X. Li, N. Pan, Y. Luo, X. Wang and J. G. Hou, ACS Nano, 2011, 5, 952–958 CrossRef CAS PubMed.
- X. Ling, L. Xie, Y. Fang, H. Xu, H. Zhang, J. Kong, M. S. Dresselhaus, J. Zhang and Z. Liu, Nano Lett., 2009, 10, 553–561 CrossRef PubMed.
- W. Ren, Y. Fang and E. Wang, ACS Nano, 2011, 5, 6425–6433 CrossRef CAS PubMed.
- Z. Zhang, F. Xu, W. Yang, M. Guo, X. Wang, B. Zhang and J. Tang, Chem. Commun., 2011, 47, 6440–6442 RSC.
- A. Saha, S. Palmal and N. R. Jana, Nanoscale, 2012, 4, 6649–6657 RSC.
- Z. Qian, Y. Cheng, X. Zhou, J. Wu and G. Xu, J. Colloid Interface Sci., 2013, 397, 103–107 CrossRef CAS PubMed.
- Y.-T. Li, L.-L. Qu, D.-W. Li, Q.-X. Song, F. Fathi and Y.-T. Long, Biosens. Bioelectron., 2013, 43, 94–100 CrossRef CAS PubMed.
- S. V. Kumar, N. M. Huang, H. N. Lim, M. Zainy, I. Harrison and C. H. Chia, Sens. Actuators, B, 2013, 181, 885–893 CrossRef CAS PubMed.
- W. Fan, Y. H. Lee, S. Pedireddy, Q. Zhang, T. Liu and X. Y. Ling, Nanoscale, 2014, 6, 4843–4851 RSC.
- D. Lin, T. Qin, Y. Wang, X. Sun and L. Chen, ACS Appl. Mater. Interfaces, 2013, 6, 1320–1329 Search PubMed.
- X. Wang, G. Meng, C. Zhu, Z. Huang, Y. Qian, K. Sun and X. Zhu, Adv. Funct. Mater., 2013, 23, 5771–5777 CrossRef CAS PubMed.
- M. Liu and W. Chen, Biosens. Bioelectron., 2013, 46, 68–73 CrossRef CAS PubMed.
- X. Liu, L. Cao, W. Song, K. Ai and L. Lu, ACS Appl. Mater. Interfaces, 2011, 3, 2944–2952 CAS.
- G. Lu, H. Li, C. Liusman, Z. Yin, S. Wu and H. Zhang, Chem. Sci., 2011, 2, 1817–1821 RSC.
- C. Hu, Y. Liu, J. Qin, G. Nie, B. Lei, Y. Xiao, M. Zheng and J. Rong, ACS Appl. Mater. Interfaces, 2013, 5, 4760–4768 CAS.
- S. Murphy, L. Huang and P. V. Kamat, J. Phys. Chem. C, 2013, 117, 4740–4747 CAS.
- L. Zhang, C. Jiang and Z. Zhang, Nanoscale, 2013, 5, 3773–3779 RSC.
- X. Li, W. C. H. Choy, X. Ren, D. Zhang and H. Lu, Adv. Funct. Mater., 2014, 24, 3114–3122 CrossRef CAS PubMed.
- P. Wang, O. Liang, W. Zhang, T. Schroeder and Y.-H. Xie, Adv. Mater., 2013, 25, 4918–4924 CrossRef CAS PubMed.
- Y. Liu, Y. Hu and J. Zhang, J. Phys. Chem. C, 2014, 118, 8993–8998 CAS.
- X. Li, J. Li, X. Zhou, Y. Ma, Z. Zheng, X. Duan and Y. Qu, Carbon, 2014, 66, 713–719 CrossRef CAS PubMed.
- Y.-K. Kim, S. W. Han and D.-H. Min, ACS Appl. Mater. Interfaces, 2012, 4, 6545–6551 CAS.
- M. Losurdo, I. Bergmair, B. Dastmalchi, T.-H. Kim, M. M. Giangregroio, W. Jiao, G. V. Bianco, A. S. Brown, K. Hingerl and G. Bruno, Adv. Funct. Mater., 2014, 24, 1864–1878 CrossRef CAS PubMed.
- Z.-L. Song, Z. Chen, X. Bian, L.-Y. Zhou, D. Ding, H. Liang, Y.-X. Zou, S.-S. Wang, L. Chen, C. Yang, X.-B. Zhang and W. Tan, J. Am. Chem. Soc., 2014, 136, 13558–13561 CrossRef CAS PubMed.
- N. Gao, Y. Chen and J. Jiang, ACS Appl. Mater. Interfaces, 2013, 5, 11307–11314 CAS.
- N. I. Kovtyukhova, P. J. Ollivier, B. R. Martin, T. E. Mallouk, S. A. Chizhik, E. V. Buzaneva and A. D. Gorchinskiy, Chem. Mater., 1999, 11, 771–778 CrossRef CAS.
- J. Zhang, J. Liu, S. Wang, P. Zhan, Z. Wang and N. Ming, Adv. Funct. Mater., 2004, 14, 1089–1096 CrossRef CAS PubMed.
- T. Liu, D. Li, D. Yang and M. Jiang, Colloids Surf., A, 2011, 387, 17–22 CrossRef CAS PubMed.
- J. Jiang, K. Bosnick, M. Maillard and L. Brus, J. Phys. Chem. B, 2003, 107, 9964–9972 CrossRef CAS.
- Y.-F. Huang, D.-Y. Wu, H.-P. Zhu, L.-B. Zhao, G.-K. Liu, B. Ren and Z.-Q. Tian, Phys. Chem. Chem. Phys., 2012, 14, 8485–8497 RSC.
- Y. Borodko, S. E. Habas, M. Koebel, P. Yang, H. Frei and G. A. Somorjai, J. Phys. Chem. B, 2006, 110, 23052–23059 CrossRef CAS PubMed.
- M. Osawa, N. Matsuda, K. Yoshii and I. Uchida, J. Phys. Chem., 1994, 98, 12702–12707 CrossRef CAS.
- H. Liang, Z. Li, W. Wang, Y. Wu and H. Xu, Adv. Mater., 2009, 21, 4614–4618 CrossRef CAS PubMed.
- G. Goncalves, S. M. A. Cruz, A. Ramalho, J. Gracio and P. A. A. P. Marques, Nanoscale, 2012, 4, 2937–2945 RSC.
- B. Han, N. Choi, K. H. Kim, D. W. Lim and J. Choo, J. Phys. Chem. C, 2011, 115, 6290–6296 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures including TEM image of SiO2@Ag without addition of PVP, zeta potential measurements, bulk Raman spectra of 4-ATP, SiO2@Ag, and SiO2@Ag@GO, 4-ATP UV-vis absorption spectra, CV and RhB SERS, GO thickness effect, and reproducibility test of the SERS substrates. See DOI: 10.1039/c5ra08180g |
| This journal is © The Royal Society of Chemistry 2015 |