DOI:
10.1039/C5RA08160B
(Paper)
RSC Adv., 2015,
5, 50691-50700
Dy/chitosan: a highly efficient and recyclable heterogeneous nano catalyst for the synthesis of hexahydropyrimidines in aqueous media†
Received
3rd May 2015
, Accepted 29th May 2015
First published on 29th May 2015
Abstract
A chitosan supported Dy(III) nanocatalyst was synthesised by a simple procedure. It was used as a catalyst for one pot, three component synthesis of hexahydropyrimidine derivatives. All the reactions were completed in a short time period at room temperature and the products were obtained in high to excellent yields. The catalyst was characterized by FT-IR, XRD, SEM-EDX, TEM and ICP-AES analyses. The stability of the catalyst was evaluated by TG analysis. Use of water, ambient conditions, recyclability and high TOF of the catalyst make this protocol a sustainable alternative to existing protocols.
Introduction
Hexahydropyrimidines are an important class of compounds with a diverse range of biological activities.1 These scaffolds are one of the most commonly encountered heterocycles in medicinal chemistry with various profiles such as antibacterial,2 antiviral, antitumor and anti-inflammatory activities.3 This skeleton is also found in a number of alkaloids such as verbamethine4 and verbametrine.5 Hexetidine, which is a hexahydropyrimidine based drug molecule, has promising anesthetic, deodorant and antiplaque effects.6 N-substituted hexahydropyrimidines serve as key synthetic intermediates for spermidine–nitroimidazole drugs which are used for the treatment of A549 lung carcinoma.7 New trypanothione reductase inhibiting ligands used for the regulation of oxidative stress in parasite cells also contain this structural unit.8 Recently, appropriately substituted hexahydropyrimidines were found to be potent hepatitis C virus inhibitors.9
Recently, lanthanides have found widespread use in the development of green chemistry as mild and efficient Lewis acids.10 A prominent feature of lanthanides is their stability and activity in protic media, making them ideal for use as stable Lewis acids in water. Dysprosium(III) is an extremely mild and efficient Lewis acid catalyst having the ability to promote various types of carbon–carbon bond-forming reactions, electrocyclizations and cycloaddition reactions.11 Compared to other lanthanides, dysprosium has not received much attention from the synthetic community even though it exhibits similar stability towards air and water, ease of handling, Lewis acidity and oxophilicity. It also has the unique ability to retain its catalytic activity in presence of Lewis basic nitrogen groups, which allows its use in a variety of transformations with unprotected amines.11
Chitosan is a naturally occurring and versatile hydrophilic polysaccharide derived from chitin. The high density of amino and hydroxyl groups of chitosan enables an effective functionalization and avoids the aggregation of metallic nanoparticles.12–16 Therefore, its use as a biodegradable supporting material for various catalysts is quite promising.
With the emphasis on the use of cleaner processes and concerns over the environmental impact of using volatile organic solvents (VOCs), increasing attention has been focused towards the use of water as reaction medium in organic syntheses. Recent research has shown that the speed of heterogeneous mixture of reactants and water is dramatically faster than for aqueous solutions. Under these heterogeneous conditions the water plays the role of a medium and not of solvent, and hence are termed as on-water reactions.17
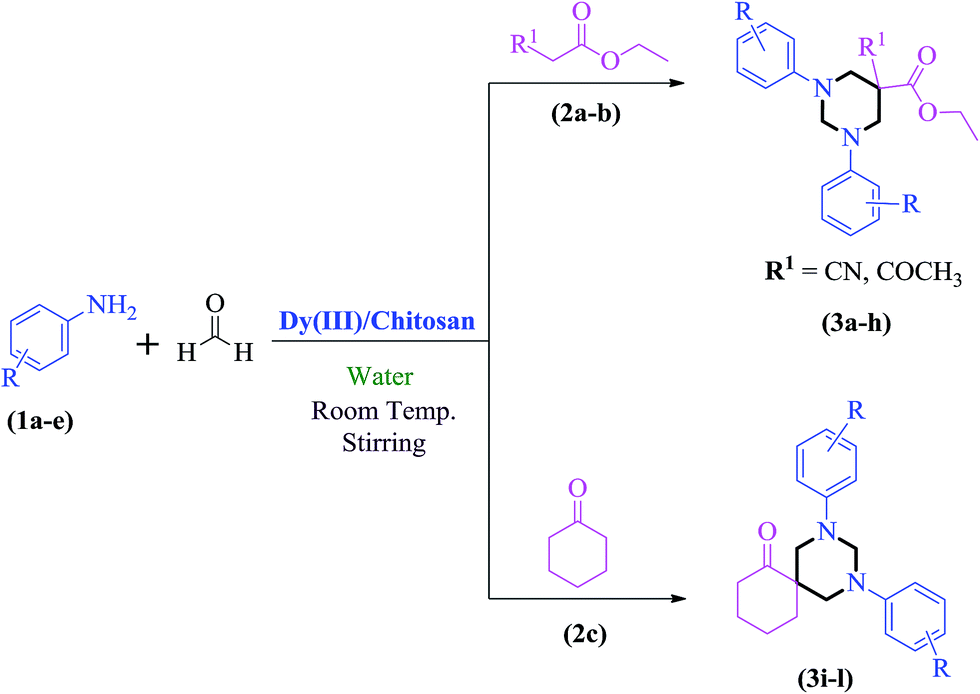
Therefore, in continuation of our interest in developing water compatible processes,18 we herein, report the synthesis of novel Dy(III)/chitosan and its application as catalyst for the synthesis of hexahydropyrimidines and their spiro analogues in water.
Results and discussion
Scheme 1 illustrates the synthesis of the catalyst. Weighed amount of chitosan was first suspended in water and stirred for 30 min. Dy(NO3)3·6H2O was then added to the suspension with continuous stirring. The mixture was then continuously stirred overnight and the catalyst was obtained by simple filtration.
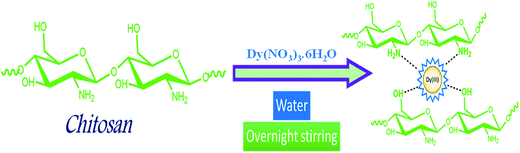 |
| | Scheme 1 Schematic representation for the synthesis of Dy(III)/chitosan catalyst. | |
Catalyst characterization
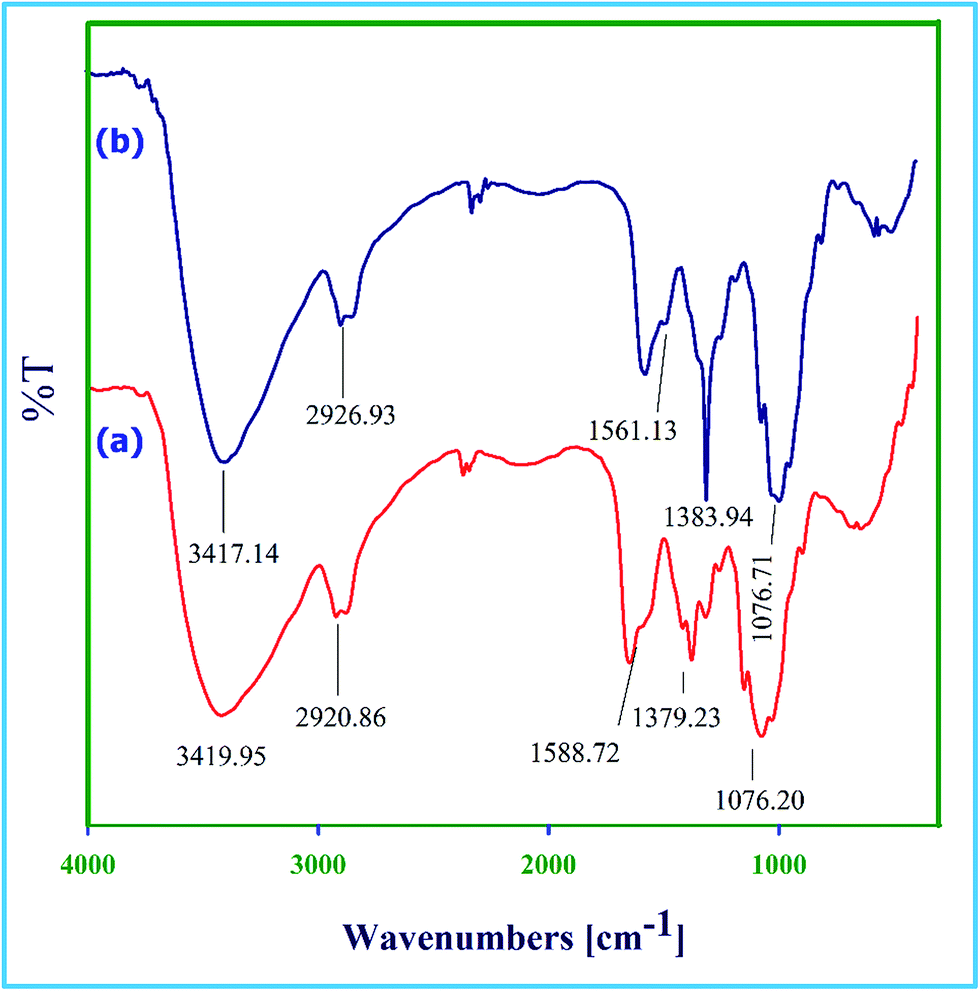
FT-IR spectra (Fig. 1a) of chitosan showed broad stretching band for OH and NH groups at 3419 cm−1. The band at 2920 cm−1 was attributed to the C–H stretching vibration of methylene groups. The band at 1588 cm−1 was assigned to N–H bending vibration of NH2 groups. The absorption bands at 1076 and 1379 cm−1 corresponded to the stretching vibrations of C–OH and C–N groups, respectively.19 The FT-IR spectra of Dy(III) supported chitosan (Fig. 1b) showed similar fingerprints compared to that of pure chitosan. The intensity of N–H bending vibration band of NH2 group at 1561 cm−1 decreased after modification, with shift in the frequency to lower wave number, indicating the coordination of Dy(III) by NH2 groups. However, due to coordination of Dy(III) by OH groups of chitosan the band at 3417 cm−1 sharpens.16
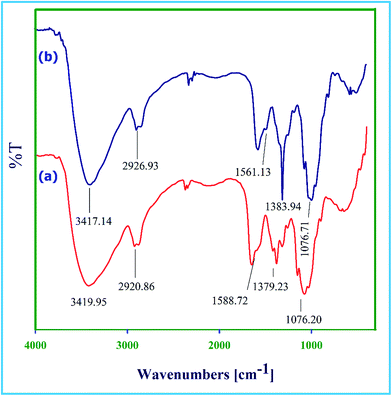 |
| | Fig. 1 FT-IR spectra (a) of chitosan and (b) of Dy(III)/chitosan catalyst. | |
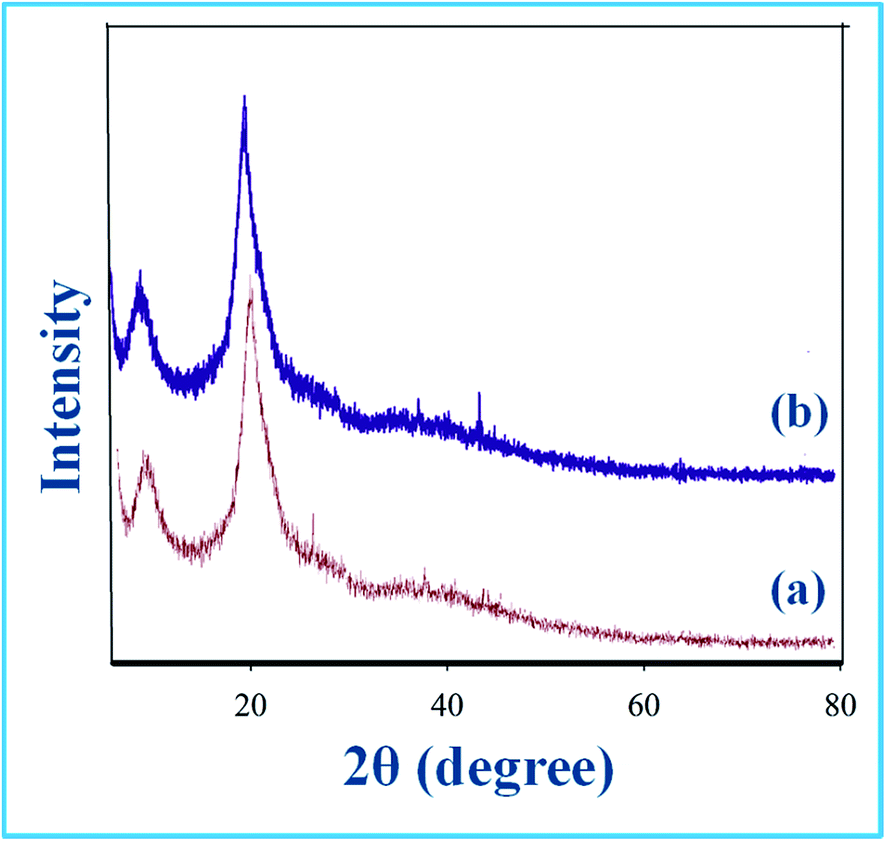
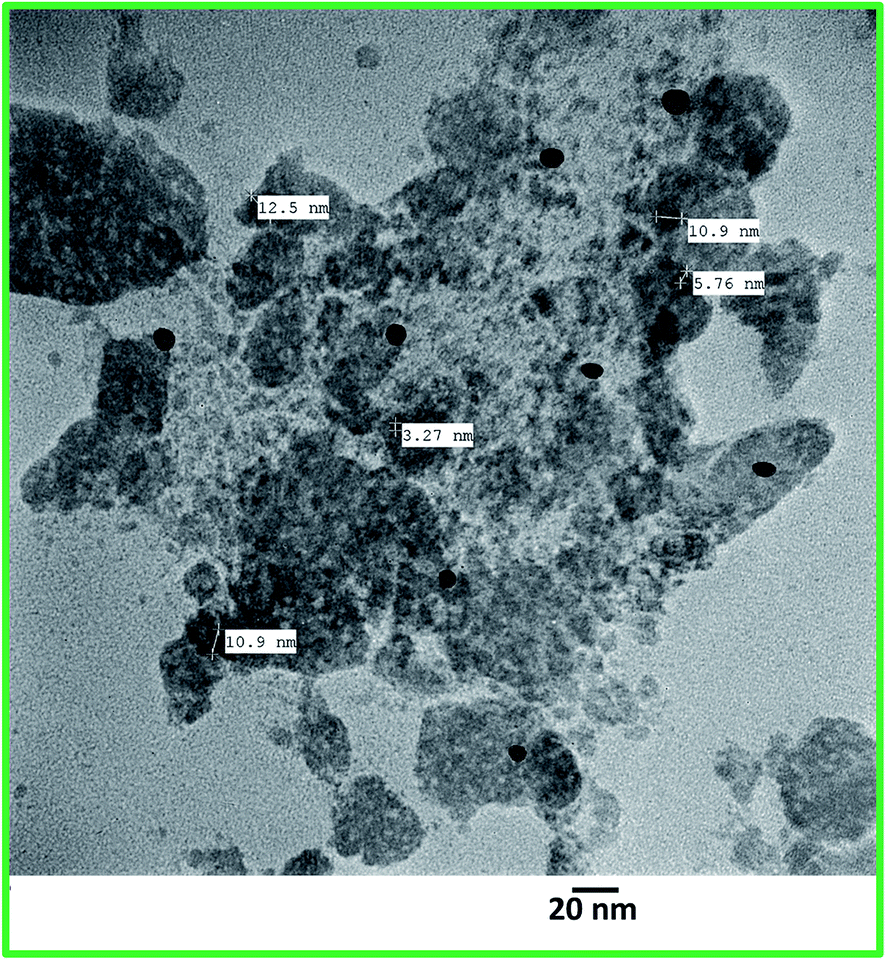
The XRD patterns of the catalyst (Fig. 2b) were similar to that of support (Fig. 2a). The peaks for the support as well as the catalyst obtained in the range 2θ (20–30°) were due to chitosan. Absence of the peaks due to Dy(III) could be because of low content and small size of the Dy(III) nanoparticles. However, the presence of Dy in the catalyst was ascertained by EDX analysis which showed peaks for the presence of Dy in addition to C, N and O elements (Fig. 3). The TEM images of the catalyst (Fig. 4) clearly displayed the spherical shape of the Dy(III) nanoparticles with average diameter in the range of 5–12 nm. The elemental mapping analysis showed the uniform distribution of Dy(III) on the surface of chitosan (Fig. 5). The surface morphology observed by SEM images at different magnifications showed the layered surface of the catalyst (Fig. 6).
 |
| | Fig. 2 XRD spectra (a) of chitosan and (b) of Dy(III)/chitosan catalyst. | |
 |
| | Fig. 3 EDX spectra of Dy(III)/chitosan catalyst. | |
 |
| | Fig. 4 TEM image of Dy(III)/chitosan catalyst. | |
 |
| | Fig. 5 Elemental mapping analysis of the catalyst showing uniform distribution of Dy. | |
 |
| | Fig. 6 SEM images of Dy(III)/chitosan catalyst at different magnifications. | |
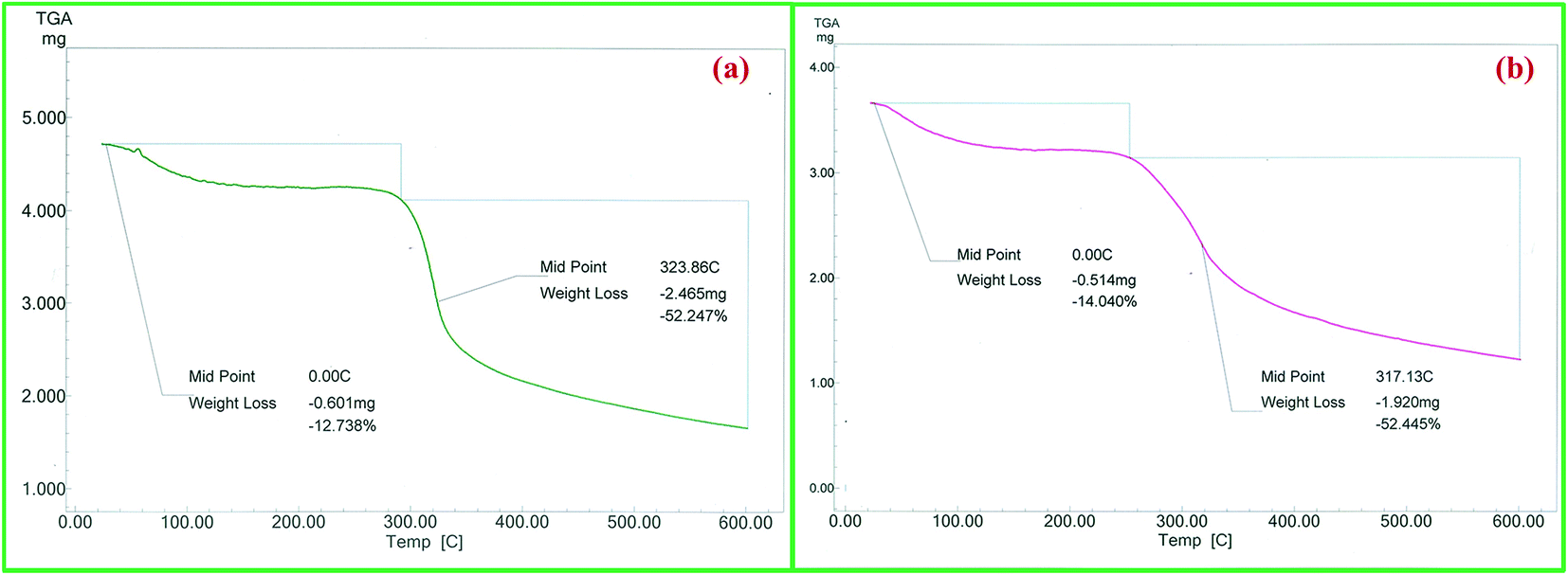
TG analysis was performed in order to evaluate the thermal behaviour and stability of the catalyst at elevated temperatures. The TG curve of chitosan showed two stage weight losses with one starting at 60 °C and another at 280 °C (Fig. 7a). The first weight loss of 12.7% is attributed to removal of adsorbed water molecules from polymer matrix and the second weight loss of 52.2% is due to the decomposition of the polysaccharide chain. Dy(III)/chitosan showed similar thermal behaviour to that of chitosan but with decrease in thermal stability (weight loss of 52.4% at 260 °C) (Fig. 7b). It is reported that chemical modifications as well as metal complexations generally lead to a decrease in the thermal stability of chitosan.20 This observation further supports the formation of Dy(III)/chitosan catalyst.
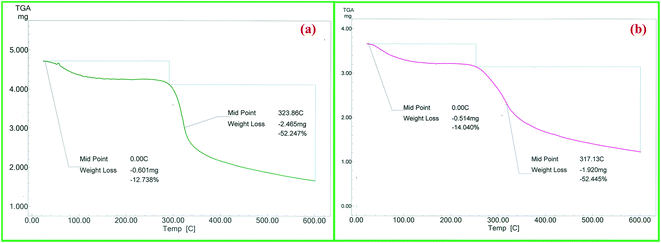 |
| | Fig. 7 TG analysis (a) of pure chitosan and (b) of Dy(III)/chitosan catalyst. | |
Optimization of reaction conditions
To investigate the feasibility of our “on-water” protocol for the synthesis of hexahydropyrimidines, sequence of experiments were carried out using aniline, formaldehyde and ethylcyano acetate as model substrates. First, various control experiments were performed, first without any catalyst and subsequently using chitosan, Dy(NO3)3·6H2O and other Lewis acids such as CuCl2, Fe(NO3)3, Zn(OAc)2 and CeCl3 as catalysts in water at room temperature (Table 1). The blank experiment without any catalyst and using chitosan as a catalyst could not show promising results (Table 1, entries 1 & 7). Among various Lewis acids, Dy(NO3)3·6H2O showed most promising catalytic effects (Table 1, entries 2–6), but its inability to be recovered and recycled led us to explore the use of chitosan as a support for the dispersion of catalytically active Dy(III). To our delight, the results obtained were very satisfactory as the product in excellent yield (91%) was obtained in short time period (Table 1, entry 8). Taking Dy(III)/chitosan as the right catalyst for this reaction, the reaction conditions were further optimized by examining the solvent effect on the reaction. Various organic solvents like ethanol, methanol, isopropanol, acetonitrile and ethylene glycol were screened to test the efficiency of the catalyst and the results are summarized in Table 1 (entries 9–13). All the solvents screened gave low to moderate yields of the product and took long time period (5–6 h) for completion of the reaction. In contrast, water showed significant improvement over other solvents in terms of yield and reaction time (Table 1, entry 8). This rate enhancement of the reaction can be attributed to the unique properties of water like hydrogen bonding to stabilize the transition states and hydrophobic effects to decrease the hydrocarbon–water interfacial area.17
Table 1 Effect of different reaction media on model reactiona
| Entry |
Catalyst |
Solvent |
Timeb |
Yieldc (%) |
| Reaction of aniline (2 mmol), ethylcyano acetate (1 mmol) and formaldehyde (3 mmol, 37–41% aqueous solution) under different conditions at room temperature. Reaction progress monitored by TLC. Isolated yield. |
| 1 |
— |
Water (5 mL) |
12 h |
— |
| 2 |
CuCl2 (10 mol%) |
Water (5 mL) |
3.1 h |
58 |
| 3 |
Fe(NO3)3 (10 mol%) |
Water (5 mL) |
2.8 h |
62 |
| 4 |
Zn(OAc)2 (10 mol%) |
Water (5 mL) |
2.5 h |
60 |
| 5 |
CeCl3 (10 mol%) |
Water (5 mL) |
2.1 h |
69 |
| 6 |
Dy(NO3)3·6H2O (10 mol%) |
Water (5 mL) |
1.6 h |
78 |
| 7 |
Chitosan (100 mg) |
Water (5 mL) |
9.5 h |
23 |
| 8 |
Dy(III)/chitosan (100 mg) |
Water (5 mL) |
45 min |
91 |
| 9 |
Dy(III)/chitosan (100 mg) |
Ethanol (5 mL) |
6 h |
61 |
| 10 |
Dy(III)/chitosan (100 mg) |
Methanol (5 mL) |
6.1 h |
64 |
| 11 |
Dy(III)/chitosan (100 mg) |
Isopropanol (5 mL) |
6.5 |
62 |
| 12 |
Dy(III)/chitosan (100 mg) |
Acetonitrile (5 mL) |
5 h |
57 |
| 13 |
Dy(III)/chitosan (100 mg) |
Ethylene glycol (5 mL) |
5.4 h |
39 |
In order to find the optimized amount of the catalyst, the reaction was carried out by varying the amount of the catalyst on the model reaction (Table 2). It was found that the conversion of the hexahydropyrimidine derivative increased linearly with increase in the amount of catalyst from 50–100 mg. Further increase in the amount of catalyst did not have any profound effect on the reaction. Therefore, 100 mg of Dy(III)/chitosan was used for the synthesis of hexahydropyrimidines.
Table 2 Effect of catalyst loadinga
| Entry |
Catalyst loading (mg) |
Timeb (min) |
Yieldc (%) |
| Reaction of aniline (2 mmol), ethylcyano acetate (1 mmol), formaldehyde (3 mmol, 37–41% aqueous solution) and different amounts of Dy(III)/chitosan in water (5 mL) at room temperature. Reaction progress was monitored by TLC. Isolated yield. |
| 1 |
50 |
125 |
69 |
| 2 |
75 |
65 |
76 |
| 3 |
100 |
45 |
91 |
| 4 |
125 |
45 |
91 |
Catalytic reaction
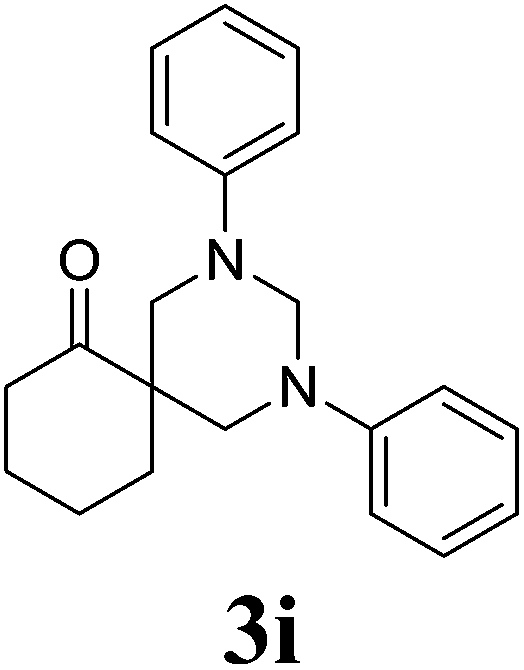
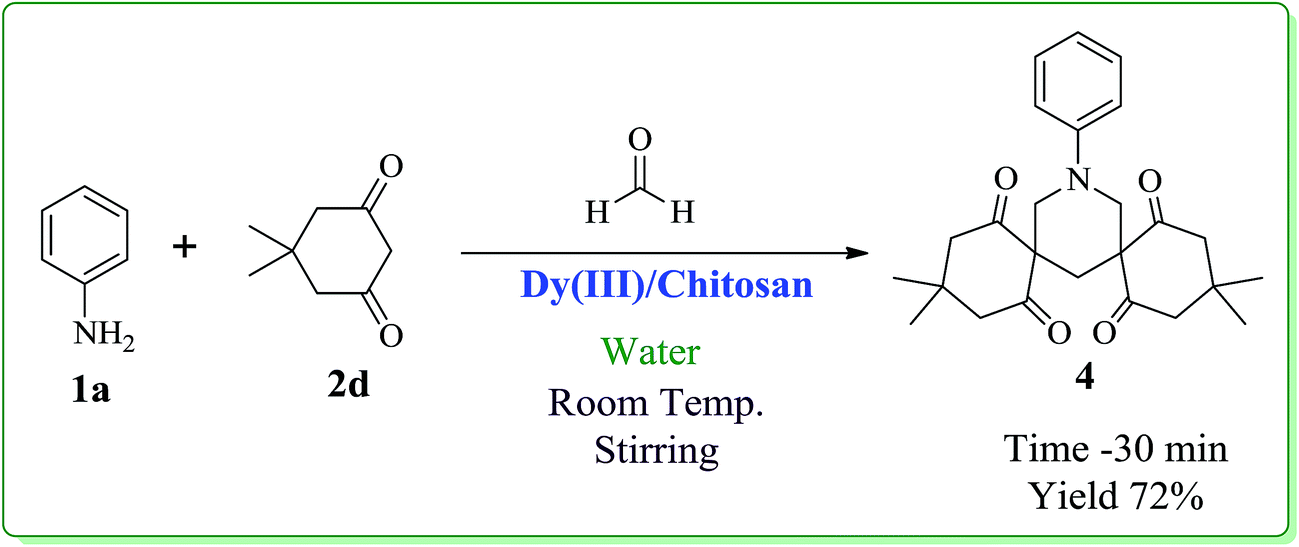
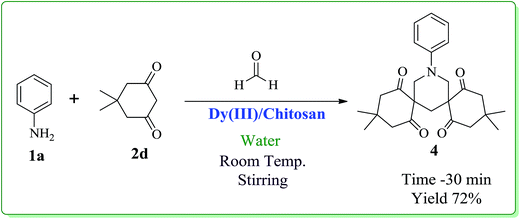
After optimizing the reaction conditions, we next proceeded to demonstrate the scope and generality of our catalytic system by applying the optimized reaction conditions for the synthesis of hexahydropyrimidine derivatives (3a–l) using different amines (1a–e), formaldehyde and active methylene compounds (2a–c) in water (Scheme 2). The results indicated that both active methylene compounds (acyclic and cyclic) and amines with activating and deactivating groups led to successful transformation to their corresponding hexahydropyrimidines (Table 3). The structures of the final products were well characterized by using spectral (IR, 1H, 13C NMR and ESI-MS) and elemental analysis data. I.R. spectrum of 3a showed a peak at 2251 cm−1 which was assigned to CN stretching. The absorption band at 1738 cm−1 was assigned to carbonyl group of ethyl ester. The 1H-NMR spectrum showed two distinctive doublets for four hydrogens of 2 × N–CH2 moieties at δ 3.63 and δ 3.77, respectively. The distinctive doublets for two hydrogen atoms of 2 × N–CH–N moieties were obtained at δ 4.24 and δ 4.74, respectively. Ten aromatic protons appeared as multiplet in the range δ 6.91–7.14. 13C-NMR showed a distinctive peak at δ 164.39 ppm for carbonyl group of ethyl ester. All other peaks were obtained at respective places and are given in the experimental section. Further, structure 3a was confirmed by ESI-Mass spectrum which showed the molecular ion peak as base peak at m/z 336.1 (M+ + 1). It is pertinent to mention that no product formation was observed when aliphatic amines (ethyl amine) or cyclic active methylene compounds (Meldrum's acid, 4-hydroxycoumarin, 1,3-dimethylbarbituric acid) were used as substrates whereas, the use of dimedone in combination with aromatic amines and formaldehyde led to the formation of spiropiperidine derivatives (Scheme 3).21 In order to show the superiority of our protocol, a comparison with reported protocols in the literature was done (Table 4). In comparison to reported procedures, our catalytic system was found to be more efficient in terms of time period and product yield.
 |
| | Scheme 2 General scheme for the synthesis of hexahydropyrimidine derivatives. | |
Table 3 Synthesis of hexahydropyrimidines and spiro analogues
| Entry |
1a–e |
2a–c |
Product |
Timea (min) |
Yieldb (%) |
TOFd |
| Reaction progress was monitored by TLC. Isolated yield. Compounds characterised on the basis of melting points, NMR and comparing them with authentic samples.21,22,24 TOF = TON/reaction time (h); TON = no. of moles of the starting materials being converted per mole of active site of the catalyst. |
| 1 |
 |
 |
 |
45 |
91 |
142.73 |
| 2 |
 |
 |
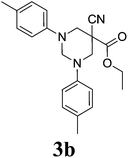 |
40 |
92 |
163.99 |
| 3 |
 |
 |
 |
45 |
90 |
141.17 |
| 4 |
 |
 |
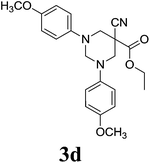 |
43 |
93 |
152.80 |
| 5 |
 |
 |
 |
48 |
89 |
130.88 |
| 6c |
 |
 |
 |
58 |
93 |
113.26 |
| 7c |
 |
 |
 |
55 |
94 |
120.72 |
| 8c |
 |
 |
 |
54 |
93 |
121.56 |
| 9c |
 |
 |
 |
50 |
87 |
123.31 |
| 10c |
 |
 |
 |
45 |
89 |
139.60 |
| 11c |
 |
 |
 |
51 |
87 |
120.41 |
| 12 |
 |
 |
 |
53 |
85 |
113.64 |
 |
| | Scheme 3 Synthesis of spiropiperidine derivative. | |
Table 4 Comparison of efficiency of Dy(III)/chitosan catalyst with reported procedures
| Entry |
Catalyst |
Condition |
Solvent |
Yield (%) |
Time |
Reference |
| 1 |
Dy(III)/chitosan |
RT stirring |
Water |
91 |
45 min |
Present work |
| 2 |
In(OTf)3 |
RT stirring |
DCM |
78 |
4 h |
22 |
| 3 |
CuFe2O4 |
RT stirring |
Ethanol |
74 |
4 h |
23 |
| 4 |
S-Proline |
RT stirring |
DMSO |
73 |
30 h |
24 |
| 5 |
(H14[NaP5W30O110])/SiO2 |
Reflux |
DMSO |
60 |
30 h |
25 |
Reaction mechanism
The mechanism of the reaction was proposed on the basis of reported literature21–24 and is outlined in Scheme 4. The catalyst first activates the active methylene compound (2a) which reacts with imine A via Michael addition to form B. The intermediate B is again activated by the catalyst and reacts with second molecule of imine A via Michael addition to form propane-1,3-diamine intermediate C. The condensation of intermediate C with formaldehyde furnishes the final product (3a).
 |
| | Scheme 4 Plausible reaction mechanism for the formation of hexahydropyrimidine derivative 3a. | |
Leaching study of Dy(III)/chitosan catalyst
The metal leaching test of the catalyst before and after six catalytic cycles was performed in order to understand the heterogeneous nature of the catalyst. ICP-AES analysis of the catalyst before (1.38 wt% Dy) and after recycling experiments (1.36 wt% Dy) revealed that the metal concentration remained unchanged with a very marginal reduction (within the experimental error of ICP-AES analysis), which in turn denoted that the metal is tightly bound to the support and no leaching of the Dy(III) occurs during reuse of the catalyst. The morphology of the catalyst, observed by TEM images, after six cycles (Fig. 8) also does not show any significant changes, indicating that the structural integrity of the catalyst is maintained throughout the recycling processes. The above studies thus, confirm that the structure of the catalyst is stable and Dy(III) is tightly bound to the support.
 |
| | Fig. 8 TEM image of recycled catalyst after six cycles. | |
Catalyst recycling
From the environmental and economic viewpoint, efficient recovery of the catalyst from the reaction mixture is the key factor that determines its utility for practical applications. Therefore, catalyst recycling experiments were employed in order to explore the extent of recyclability of our catalytic system. The reaction between aniline, formaldehyde (37–41% aqueous solution) and ethylcyano acetate in water at room temperature using Dy(III)/chitosan as a catalyst was chosen as model reaction. After completion of the reaction, the catalyst was recovered by extracting the mixture with ethyl acetate followed by filtration. The catalyst was then washed with ethyl acetate and reused for subsequent cycles. The catalyst was found to retain its activity for a minimum of six reaction cycles in water displaying a high catalytic performance with over 83% yield of the product (Table 5).
Table 5 Recycling potential of Dy(III)/chitosan catalysta
| No. of cycles |
Fresh |
Run 1 |
Run 2 |
Run 3 |
Run 4 |
Run 5 |
| Reaction of aniline (2 mmol), ethylcyano acetate (1 mmol), formaldehyde (3 mmol, 37–41% aqueous solution) and Dy(III)/chitosan (100 mg) in water (5 mL) at room temperature. |
| Yield (%) |
91 |
91 |
89 |
86 |
85 |
83 |
| Time (min) |
45 |
45 |
45 |
45 |
45 |
45 |
| TOF |
142.73 |
142.73 |
139.60 |
134.90 |
133.33 |
130.20 |
Experimental
General
Melting points of all the synthesized compounds were taken in a Riechert Thermover instrument and are uncorrected. The FT-IR spectra were recorded on Perkin Elmer RXI spectrometer using KBr pellets. 1H NMR and 13C NMR spectra were recorded on a Bruker DRX-400 spectrometer using tetramethylsilane (TMS) as an internal standard and CDCl3 as solvent. Mass spectra were recorded on Micromass Quattro II (ESI) spectrometer. Elemental analyses (C, H and N) were conducted using the Elemental vario EL III elemental analyser and their results were found to be in agreement with the calculated values. X-ray diffractograms (XRD) of the catalyst were recorded in the 2θ range of 5–80° with scan rate of 4° min−1 on a Rigaku Minifax X-ray diffractometer with Ni-filtered Cu Kα radiation at a wavelength of 1.54060 Å. The SEM and EDX characterization of the catalyst was performed on JEOL JSM-6510 scanning electron microscope equipped with energy dispersive X-ray spectrometer operating at 20 kV. TEM analysis was performed on JEM-2100 F Model (ACC. voltage: 200 kV) electron microscope. ICP-AES analysis was performed on ARCOS from M/s. Spectro, Germany. All reagents were purchased from Merck and Aldrich and were used without further purification. The purity of compounds was checked by thin layer chromatography (TLC) on glass plates coated with silica gel G254 (E. Merck) using chloroform–methanol (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture as mobile phase and visualised by iodine vapours and alcoholic ferric chloride.
:1) mixture as mobile phase and visualised by iodine vapours and alcoholic ferric chloride.
Synthesis of Dy(III)/chitosan catalyst. Chitosan (3 g) was suspended in 100 mL of distilled water in a round bottomed flask and stirred for 30 min. Dy(NO3)3·6H2O (0.2 g) was then added to the suspension with continuous stirring. The mixture was then continuously stirred overnight and the solid catalyst was separated by filtration, washed with water and dried at 80 °C for 6 h.
General procedure for the synthesis of hexahydropyrimidine derivatives. A mixture of amine (1a–e) (2 mmol), active methylene compound (2a–c) (1 mmol), formaldehyde (3 mmol, 37–41% aqueous solution) and catalyst (100 mg) in 5 mL water was stirred at room temperature for appropriate period of time (Table 3). After completion of reaction (monitored by TLC), the reaction mixture was allowed to cool, extracted by ethyl acetate, washed with water (3 × 10 mL), dried over anhydrous Na2SO4 and evaporated under reduced pressure to obtain the products (3a–e and 3i–l). The remaining solid catalyst in aqueous phase was separated by filtration, washed with ethyl acetate (3 × 10 mL) and reused for further catalytic cycles. The crude products were recrystallized to afford the pure products. Compounds (3f–h) were separated using silica gel (60–120 mesh) column chromatography with 5% ethyl acetate in petroleum ether as eluent.
Spectral data of synthesised compounds
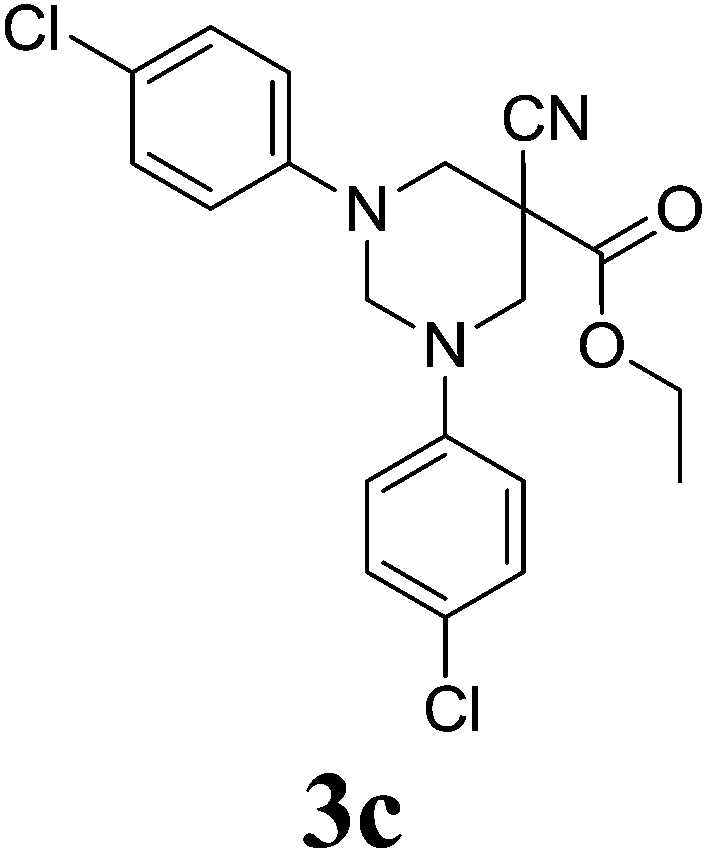
5-Cyano-1,3-diphenyl-hexahydro-pyrimidine-5-carboxylic acid ethyl ester 3a. Mp 120–125 °C; IR (KBr, cm−1): 2251 (CN), 1738 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O). 1H NMR (400 MHz, CDCl3): δ 1.16 (s, 3H), 3.63 (d, 2H, N–CH2), 3.77 (d, 2H, N–CH2), 4.09 (q, 2H), 4.24 (d, 1H, N–CH–N), 4.74 (d, 1H, N–CH–N), 6.91–7.14 (m, 10H, Ar-region). 13C NMR (100 MHz, CDCl3): 164.39, 149.27, 131.54, 127.29, 121.22, 117.13, 70.25, 64.81, 62.18, 52.23, 14.18. Anal. calcd C, 71.62; H, 6.31; N, 12.53; anal. found C, 71.66; H, 6.28; N, 12.57. ESI-MS m/z: 336.1 (M+ + 1).
O). 1H NMR (400 MHz, CDCl3): δ 1.16 (s, 3H), 3.63 (d, 2H, N–CH2), 3.77 (d, 2H, N–CH2), 4.09 (q, 2H), 4.24 (d, 1H, N–CH–N), 4.74 (d, 1H, N–CH–N), 6.91–7.14 (m, 10H, Ar-region). 13C NMR (100 MHz, CDCl3): 164.39, 149.27, 131.54, 127.29, 121.22, 117.13, 70.25, 64.81, 62.18, 52.23, 14.18. Anal. calcd C, 71.62; H, 6.31; N, 12.53; anal. found C, 71.66; H, 6.28; N, 12.57. ESI-MS m/z: 336.1 (M+ + 1).
5-Cyano-1,3-di-(4-methylphenyl)-hexahydro-pyrimidine-5-carboxylic acid ethyl ester 3b. Mp 115–120 °C; IR (KBr, cm−1): 2244 (CN), 1729 (CO). 1H NMR (400 MHz, CDCl3): δ 1.22 (s, 3H), 2.29 (s, 6H), 3.71 (d, 2H, N–CH2), 3.90 (d, 2H, N–CH2), 4.12 (q, 2H), 4.27 (d, 1H, N–CH–N), 4.72 (d, 1H, N–CH–N), 6.97–7.12 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 166.14, 148.10, 131.27, 127.01, 122.75, 118.52, 69.07, 64.22, 61.59, 51.88, 21.35, 14.86. Anal. calcd C, 72.70; H, 6.93; N, 11.56; anal. found C, 72.75; H, 6.89; N, 11.52. ESI-MS m/z: 364.1 (M+ + 1).
5-Cyano-1,3-di-(4-chlorophenyl)-hexahydro-pyrimidine-5-carboxylic acid ethyl ester 3c. Mp 110–115 °C; IR (KBr, cm−1): 2241 (CN), 1725 (CO). 1H NMR (400 MHz, CDCl3): δ 1.24 (s, 3H), 3.72 (d, 2H, N–CH2), 3.93 (d, 2H, N–CH2), 4.15 (q, 2H), 4.31 (d, 1H, N–CH–N), 4.68 (d, 1H, N–CH–N), 6.89–7.21 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 165.11, 149.19, 132.87, 129.27, 124.81, 117.95, 71.03, 64.44, 61.94, 51.76, 14.43. Anal. calcd C, 59.42; H, 4.74; N, 10.39; anal. found C, 59.46; H, 4.78; N, 10.36. ESI-MS m/z: 404.1 (M+ + 1).
5-Cyano-1,3-di-(4-methoxyphenyl)-hexahydro-pyrimidine-5-carboxylic acid ethyl ester 3d. Mp 120–125 °C; IR (KBr, cm−1): 2249 (CN), 1731 (CO). 1H NMR (400 MHz, CDCl3): δ 1.25 (s, 3H), 3.69 (d, 2H, N–CH2), 3.75 (s, 6H, 2 × OCH3), 3.87 (d, 2H, N–CH2), 4.11 (q, 2H), 4.40 (d, 1H, N–CH–N), 4.74 (d, 1H, N–CH–N), 6.88–7.25 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 168.07, 148.23, 134.42, 129.08, 124.27, 119.18, 71.13, 63.18, 62.54, 55.81, 51.88, 15.11. Anal. calcd C, 66.82; H, 6.37; N, 10.63; anal. found C, 66.78; H, 6.34; N, 10.68. ESI-MS m/z: 396.1 (M+ + 1).
5-Cyano-1,3-di-(4-nitrophenyl)-hexahydro-pyrimidine-5-carboxylic acid ethyl ester 3e. Mp 125–130 °C; IR (KBr, cm−1): 2238 (CN), 1724 (CO). 1H NMR (400 MHz, CDCl3): δ 1.27 (s, 3H), 3.76 (d, 2H, N–CH2), 3.92 (d, 2H, N–CH2), 4.14 (q, 2H), 4.28 (d, 1H, N–CH–N), 4.78 (d, 1H, N–CH–N), 7.14–8.26 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 167.12, 149.22, 135.82, 131.21, 125.03, 117.73, 72.02, 64.11, 63.58, 51.97, 15.77. Anal. calcd C, 56.47; H, 4.50; N, 16.46; anal. found C, 56.43; H, 4.53; N, 16.49. ESI-MS m/z: 426.1 (M+ + 1).
5-Acetyl-1,3-diphenyl-hexahydro-pyrimidine-5-carboxylic acid ethyl ester 3f21. 1H NMR (400 MHz, CDCl3): δ 1.19 (t, 3H), 2.27 (s, 3H), 3.88 (d, 2H, N–CH2), 3.97 (d, 2H, N–CH2), 4.02 (q, 2H), 4.29 (d, 1H, N–CH–N), 4.38 (d, 1H, N–CH–N), 6.79–7.31 (m, 10H, Ar-region). 13C NMR (100 MHz, CDCl3): 195.64, 164.39, 147.38, 129.52, 121.41, 117.13, 67.91, 61.39, 60.23, 50.31, 26.43, 13.24. ESI-MS m/z: 353.1 (M+ + 1).
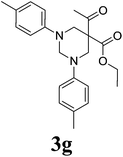
5-Acetyl-1,3-di-(4-methylphenyl)-hexahydro-pyrimidine-5-carboxylic acid ethyl ester 3g21. 1H NMR (400 MHz, CDCl3): δ 1.13 (t, 3H), 2.25 (s, 3H), 2.64 (s, 6H), 3.79 (d, 2H, N–CH2), 3.88 (d, 2H, N–CH2), 4.06 (q, 2H), 4.31 (d, 1H, N–CH–N), 4.51 (d, 1H, N–CH–N), 6.73–7.24 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 194.70, 166.23, 148.15, 130.07, 129.17, 117.49, 68.65, 61.23, 59.15, 54.37, 26.18, 20.43, 14.36. ESI-MS m/z: 381.1 (M+ + 1).
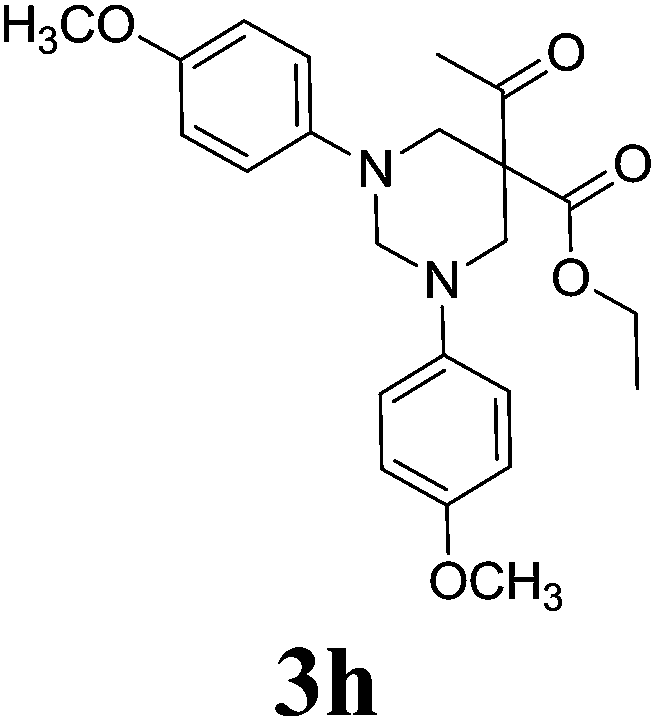
5-Acetyl-1,3-di-(4methoxyphenyl)-hexahydro-pyrimidine-5-carboxylic acid ethyl ester 3h21. 1H NMR (400 MHz, CDCl3): δ 1.11 (t, 3H), 2.28 (s, 3H), 3.71 (s, 6H), 3.84 (d, 2H, N–CH2), 3.96 (d, 2H, N–CH2), 4.13 (q, 2H), 4.33 (d, 1H, N–CH–N), 4.59 (d, 1H, N–CH–N), 7.13–7.92 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 195.82, 165.30, 148.00, 130.78, 129.27, 120.22, 68.94, 62.08, 58.23, 55.91, 54.18, 26.68, 20.37, 13.78. ESI-MS m/z: 413.1 (M+ + 1).
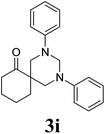
2,4-Bis-phenyl-2,4-diazaspiro[5.5]undecan-7-one 3i24. 1H NMR (400 MHz, CDCl3): δ 1.58–1.63 (m, 2H), 1.83–1.95 (m, 4H), 2.45 (t, 2H), 3.46 (d, 2H), 3.63 (d, 2H), 4.09 (d, 1H), 4.66 (d, 1H), 6.79–7.23 (m, 10H, Ar-region). 13C NMR (100 MHz, CDCl3): 198.13, 149.65, 129.33, 120.21, 118.49, 68.21, 55.11, 50.73, 39.78, 34.36, 27.73, 20.33. ESI-MS m/z: 321.1 (M+ + 1).
2,4-Bis-(4-methylphenyl)-2,4-diazaspiro[5.5]undecan-7-one 3j24. Mp 127 °C; 1H NMR (400 MHz, CDCl3): δ 1.61–1.67 (m, 2H), 1.81–1.92 (m, 4H), 2.35 (s, 6H), 2.51 (t, 2H), 3.42 (d, 2H), 3.61 (d, 2H), 4.12 (d, 1H), 4.74 (d, 1H), 6.67–7.38 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 198.68, 148.51, 129.95, 121.52, 118.11, 69.53, 55.11, 50.28, 39.16, 34.98, 27.10, 22.44, 20.13. ESI-MS m/z: 349.1 (M+ + 1).
2,4-Bis-(4-chlorophenyl)-2,4-diazaspiro[5.5]undecan-7-one 3k24. Mp 161 °C; 1H NMR (400 MHz, CDCl3): δ 1.64–1.69 (m, 2H), 1.81–1.90 (m, 4H), 2.42 (t, 2H), 3.44 (d, 2H), 3.64 (d, 2H), 4.17 (d, 1H), 4.63 (d, 1H), 6.79–7.13 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 199.57, 148.87, 128.38, 125.61, 118.51, 67.30, 56.25, 49.21, 39.37, 34.96, 27.32, 20.08. ESI-MS m/z: 389.1 (M+ + 1).
2,4-Bis-(4-nitrophenyl)-2,4-diazaspiro[5.5]undecan-7-one 3l. Mp 170–175 °C; 1H NMR (400 MHz, CDCl3): δ 1.68–1.72 (m, 2H), 1.88–1.95 (m, 4H), 2.45 (t, 2H), 3.41 (d, 2H), 3.62 (d, 2H), 4.10 (d, 1H), 4.58 (d, 1H), 7.09–8.11 (m, 8H, Ar-region). 13C NMR (100 MHz, CDCl3): 199.52, 147.73, 138.87, 124.34, 117.62, 68.01, 56.44, 49.93, 38.89, 34.54, 27.11, 20.52. ESI-MS m/z: 411.1 (M+ + 1).
3,3,11,11-Tetramethyl-15-(phenyl)-15-azadispiro[5.1.5.3]hexadecane-1,5,9,13-tetrone 4. Mp 191–193 °C; 1H NMR (400 MHz, CDCl3): δ 0.85 (s, 6H), 0.94 (s, 6H), 2.32–2.37 (m, 6H), 2.67 (d, 4H), 3.52 (3, 4H), 7.25–7.32 (m, 6H, Ar-region). 13C NMR (100 MHz, CDCl3): 198.50, 149.01, 131.27, 124.93, 119.45, 66.25, 55.33, 51.97, 32.89, 31.77, 29.08, 28.58.
Conclusion
In summary, we have synthesised a new chitosan supported Dy(III) catalyst and used it for the green and energy sustainable synthesis of hexahydropyrimidine derivatives. The catalyst is found to be highly efficient and could be reused for six catalytic cycles. The use of water, substrate tolerance, heterogeneous catalyst and ambient conditions make this protocol an energy sustainable alternative to reported methods.
Acknowledgements
The authors are thankful to CST-UP (no. CST/SERPD/D-283, Dated 14-May-2015) for financial assistance, department of physics, AMU, for XRD analysis, University Sophisticated Instrument Facility (USIF), AMU, Aligarh, for SEM-EDX and TEM facilities, SAIF, IIT Bombay for ICP-AES analysis and SAIF Punjab University, Chandigarh for providing NMR and Mass spectra. N.A. thanks UGC for providing fellowship.
References
- K. S. Jain, T. S. Chitre, P. B. Miniyar, M. K. Kathiravan, V. S. Bendre, V. S. Veer, S. R. Shahane and C. J. Shishoo, Curr. Sci., 2006, 90, 793 CAS.
- J. A. Bisceglia, M. B. Garcia, R. Massa, M. L. Magri, M. Zani, G. O. Gutkind and L. R. Orelli, J. Heterocycl. Chem., 2004, 41, 85 CrossRef CAS.
-
(a) S. Tu, C. Miao, F. Fang, F. Youjian, T. Li, Q. Zhuang, X. Zhang, S. Zhu and D. Shi, Bioorg. Med. Chem. Lett., 2004, 14, 1533 CrossRef CAS PubMed;
(b) C. O. Kappe, Tetrahedron, 1993, 49, 6937 CrossRef CAS;
(c) C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043 CrossRef CAS PubMed;
(d) D. Russowski, R. F. S. Canto, S. A. A. Sanches, M. G. M. D'Oca, A. D. Fatima and J. E. D. Carvalho, Bioorg. Chem., 2006, 34, 173 CrossRef PubMed.
- A. Guggisberg, K. Drandarov and M. Hesse, Helv. Chim. Acta, 2000, 83, 3035 CrossRef CAS.
-
(a) J. C. Braekman, D. Daloze, R. Flammang and A. Maquestiau, Org. Mass Spectrom., 1989, 24, 837 CrossRef;
(b) K. Drandarov, A. Guggisberg and M. Hesse, Helv. Chim. Acta, 1999, 82, 229 CrossRef CAS.
- P. A. S. Smith and R. N. Loeppky, J. Am. Chem. Soc., 1967, 89, 1147 CrossRef CAS.
- A. Q. Siddiqui, L. Merson-Davies and P. M. Cullis, J. Chem. Soc., Perkin Trans. 1, 1999, 1, 3243 RSC.
- D. Hrvath, J. Med. Chem., 1997, 40, 2412 CrossRef PubMed.
- J. W. Hwang, H.-W. Kim, S. Jo, E. Park, J. Choi, S. Kong, D.-S. Park, J. M. Heo, J. S. Lee, Y. Ko, I. Choi, J. Cechetto, J. Kim, J. Lee, Z. No and M. P. Windisch, Eur. J. Med. Chem., 2013, 70, 315 CrossRef CAS PubMed.
-
(a) H. B. Kagan and J. L. Namy, Tetrahedron, 1986, 42, 6573 CrossRef CAS;
(b) G. A. Molander, Chem. Rev., 1992, 92, 29 CrossRef CAS;
(c) K. Mikami, M. Terada and H. Matsuzawa, Angew. Chem., Int. Ed., 2002, 41, 3554 CrossRef CAS;
(d) S. Kobayashi, Eur. J. Org. Chem., 1999, 15 CrossRef CAS;
(e) S. Kobayashi, Top. Organomet. Chem., 1999, 2, 63 CrossRef CAS;
(f) R. Anwander, Top. Organomet. Chem., 1999, 2, 1 CrossRef CAS.
- G. K. Veits and J. R. de Alaniz, Tetrahedron, 2012, 68, 2015 CrossRef CAS.
- D. M. Whitfield, S. Stojkovski and B. Sarkar, Coord. Chem. Rev., 1993, 122, 171 CrossRef CAS.
- A. Patwardhan and J. A. Cowan, Dalton Trans., 2011, 40, 1795 RSC.
- A. Corma, P. Concepcin, I. Domnguez, V. Fornes and M. J. Sabater, J. Catal., 2007, 251, 39 CrossRef CAS.
-
(a) J. X. Jian, Q. Liu, Z. J. Li, F. Wang, X. B. Li, C. B. Li, B. Liu, Q. Y. Meng, B. Chen, K. Feng, C. H. Tung and L. Z. Wu, Nat. Commun., 2013, 4, 2695 Search PubMed;
(b) R. B. N. Baig, M. N. Nadagouda and R. S. Varma, Green Chem., 2014, 16, 2122 RSC.
-
(a) A. A. Dabbawala, N. Sudheesh and H. C. Bajaj, Dalton Trans., 2012, 41, 2910 RSC;
(b) S. E. S. Leonhardt, A. Stolle, B. Ondruschka, G. Cravotto, C. De Leo, K. D. Jandt and T. F. Keller, Appl. Catal., A, 2010, 379, 30 CrossRef CAS;
(c) C. Shen, J. Xu, W. Yu and P. Zhang, Green Chem., 2014, 16, 3007 RSC.
-
(a) J. E. Klijn and J. B. F. N. Engberts, Nature, 2005, 435, 746 CrossRef CAS PubMed;
(b) A. Manna and A. Kumar, J. Phys. Chem. A, 2013, 117, 2446 CrossRef CAS PubMed;
(c) Y. Jung and R. A. Marcus, J. Am. Chem. Soc., 2007, 129, 5492 CrossRef CAS PubMed;
(d) C.-J. Li, Chem. Rev., 2005, 105, 3095 CrossRef CAS PubMed.
-
(a) N. Ahmed and Z. N. Siddiqui, RSC Adv., 2015, 5, 16707 RSC;
(b) N. Ahmed and Z. N. Siddiqui, J. Mol. Catal. A: Chem., 2014, 394, 232 CrossRef CAS.
- N. Sudheesh, S. K. Sharma and R. S. Shukla, J. Mol. Catal. A: Chem., 2010, 321, 77 CrossRef CAS.
- K. D. Trimukhea and A. J. Varma, Carbohydr. Polym., 2009, 75, 63 CrossRef.
- C. Mukhopadhyay, S. Rana and R. J. Butcher, Tetrahedron Lett., 2011, 52, 4153 CrossRef CAS.
- A. Dandia, A. K. Jain and S. Sharma, Tetrahedron Lett., 2012, 53, 5270 CrossRef CAS.
- A. Dandia, A. K. Jain and S. Sharma, RSC Adv., 2013, 3, 2924 RSC.
- H.-L. Wei, Z.-Y. Yan, Y.-N. Niu, G.-Q. Li and Y.-M. Liang, J. Org. Chem., 2007, 72, 8600 CrossRef CAS PubMed.
- M. M. Heravi, S. Sadjadi, S. Sadjadi, H. A. Oskooie, R. H. Shoar and F. F. Bamoharram, J. Chin. Chem. Soc., 2009, 56, 246 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra08160b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.