
Dispersity control of linear poly(glycidyl ether)s by slow monomer addition†
M. Kuhlmann and
J. Groll*
Department for Functional Materials in Medicine and Dentistry, University of Würzburg, Pleicherwall 2, 97070 Würzburg, Germany. E-mail: juergen.groll@fmz.uni-wuerzburg.de
First published on 30th July 2015
Abstract
In this communication we demonstrate that the extent of dispersity of poly(allyl glycidyl ether) and poly(ethoxy ethyl glycidyl ether) can be reduced using a slow monomer addition technique with potassium tert-butoxide as initiator and THF as solvent at 45 °C. Different feed rates were examined in triplicate and analysed by SEC.
Poly(allyl glycidyl ether) (poly(AGE)) homo- and copolymers are promising alternatives to low-functional poly(ethylene oxide) (PEO).1 Ethylene oxide (EO), ethoxy ethyl glycidyl ether (EEGE) and other oxirane-monomers have already been statistically or block copolymerized with allyl glycidyl ether (AGE) to improve functionalization options.2–4 Using this approach, modification of the unsaturated side-chains (e.g. via thiol–ene click chemistry) has allowed the synthesis of multifunctional PEO-derivatives over the past decades.5–7
Side-reactions and their control in glycidyl ether polymerizations were investigated as this determines the applicability, i.e. reproducibility, control over molar mass and dispersity. Challenges in syntheses were believed to be due to isomerization reactions and transfer reactions. Depending on the media, temperature, and polymerization parameters the transfer reactions limit the degree of polymerization and increase dispersity.6,8–11 Hans et al. assumed that chain transfer occurs in EEGE polymerization by methylene-group deprotonation in α-position to the oxirane as it is known for propylene oxide (PO) and other oxiranes for decades.12 Here the newly formed allyl-alkoxide initiates a new polymerization that leads to an additional low molar-mass fraction and tailing in SEC-traces. Furthermore, at higher temperatures dimerization can occur that leads to polymer fractions with twice the molar mass of the main polymer fraction. Mechanistically, deprotonation at the oxirane-ring leads to a ketone group and a carbanionic species. This initiates a new polymerization and gives a ketone-terminal polymer that can couple with other oxyanionic species.8 The two mentioned side-reactions lead to low and high molar-mass impurities. Erberich et al. assumed that controlled polymerization of AGE proceeds only up to 80% conversion. Afterwards deprotonation reactions occur that terminate the active chain. The authors used potassium alkoxide as initiator (with only 10% of active alkoxides) at 120 °C in diglyme. The obtained poly(allyl glycidyl ether) had a DP of 21 and a dispersity of 1.27. In contrast, Lee et al. used potassium benzoxide and polymerized AGE in diglyme or neat at 30–80 °C and a controlled polymerization with DP up to 800 was possible. At higher temperatures the authors observed isomerization reactions that lead to cis-prop-1-enyl side-chains. Please note that this isomerization at the side-reaction does not terminate the polymerization.1
Caesium-alkoxides are one promising alternative to potassium-alkoxides for the polymerization of glycidyl ethers as the Cs+-alkoxides exhibit a pronounced ionic character and increase the activity of the chain ends.13 Furthermore, isomerization reactions are reduced using Cs+ as counterion enhancing the control over unwanted side-reactions.14 Unfortunately, the use of caesium-alkoxides is limited due to solubility issues of caesium-initiators in various solvents. For example, caesium 2-methoxyethoxide was used as initiator although it shows a low solubility in THF. To increase the solubility DMSO was added.6 For benzene as solvent a PEG initiator with Mn > 1000 g mol−1 is required to dissolve the initiator properly if both hydroxyl groups were activated with CsOH.15 In contrast to caesium-alkoxides, low molar-mass potassium alkoxides such as potassium ethoxide16 and potassium t-butoxide readily dissolve in THF or benzene. Furthermore, upon preparation of the caesium-alkoxides protic compounds are often required and H2O is released that has to be completely removed before polymerization.1,17,18
During the synthesis of statistical poly(EEGE-co-AGE) with [M]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[I] of 50:1 and various fractions of AGE, bimodal SEC elugrams were obtained in our labs. The bimodality was more pronounced than described for poly(EEGE) homopolymers by Hans et al.8 It is known that a long-term standing of poly(AGE) induces an uncontrolled radical process leading to cross-linking of poly(AGE).3 Bimodal SEC elugrams having one polymer fraction with the intended molar mass and one fraction having twice the molar-masses were obtained. This indicated the aforementioned ketone-coupling theory rather than an uncontrolled radical cross-linking of the unsaturated side-chains of the polymers. Dispersities and elugrams were compared with literature, but Hans et al. reported that the fraction with twice the molar mass occurs at higher temperatures for EEGE. A further look into the polymerization parameters revealed that the ratio [M]:[I] was also lower than reported here.3,8,19 Deprotonation side-reactions occur more likely with higher [M]:[I] ratios that result in a higher fraction of coupled polymers. Hence, we hypothesized that a step-wise addition of the monomer to the initiator/reaction solution keeps the monomer content low and reduces the ketone-induced coupling. Additionally, we assumed that initially formed ketone-functional polymers couple reversibly with oxyanionic species. This assumption is based on the following considerations. An irreversible dimerization of low molar-mass polymers would lead to low molar-mass fractions visible by a tailing in SEC elugrams. Monomer deprotonation reactions that occur preferably at the end of polymerization do also not result in polymer fractions with twice the molar mass. Here, the residual monomer content is not sufficiently high to increase the molar-mass of the polymers to such an extent. The polymer fraction with twice the molar-mass is therefore most probable by an initial deprotonation reaction. Concurrently with the polymerization, a reversible ketone–alkoxide coupling occurs. The equilibrium between the active (non-coupled) and dormant (coupled) species is furthermore assumed to be rapid compared to the polymerization itself. If this is not the case a trimer would be observable in SEC elugrams. The formed alkoxide-functional dimer would initiate a new polymerization (Scheme 1).
:[I] of 50:1 and various fractions of AGE, bimodal SEC elugrams were obtained in our labs. The bimodality was more pronounced than described for poly(EEGE) homopolymers by Hans et al.8 It is known that a long-term standing of poly(AGE) induces an uncontrolled radical process leading to cross-linking of poly(AGE).3 Bimodal SEC elugrams having one polymer fraction with the intended molar mass and one fraction having twice the molar-masses were obtained. This indicated the aforementioned ketone-coupling theory rather than an uncontrolled radical cross-linking of the unsaturated side-chains of the polymers. Dispersities and elugrams were compared with literature, but Hans et al. reported that the fraction with twice the molar mass occurs at higher temperatures for EEGE. A further look into the polymerization parameters revealed that the ratio [M]:[I] was also lower than reported here.3,8,19 Deprotonation side-reactions occur more likely with higher [M]:[I] ratios that result in a higher fraction of coupled polymers. Hence, we hypothesized that a step-wise addition of the monomer to the initiator/reaction solution keeps the monomer content low and reduces the ketone-induced coupling. Additionally, we assumed that initially formed ketone-functional polymers couple reversibly with oxyanionic species. This assumption is based on the following considerations. An irreversible dimerization of low molar-mass polymers would lead to low molar-mass fractions visible by a tailing in SEC elugrams. Monomer deprotonation reactions that occur preferably at the end of polymerization do also not result in polymer fractions with twice the molar mass. Here, the residual monomer content is not sufficiently high to increase the molar-mass of the polymers to such an extent. The polymer fraction with twice the molar-mass is therefore most probable by an initial deprotonation reaction. Concurrently with the polymerization, a reversible ketone–alkoxide coupling occurs. The equilibrium between the active (non-coupled) and dormant (coupled) species is furthermore assumed to be rapid compared to the polymerization itself. If this is not the case a trimer would be observable in SEC elugrams. The formed alkoxide-functional dimer would initiate a new polymerization (Scheme 1).
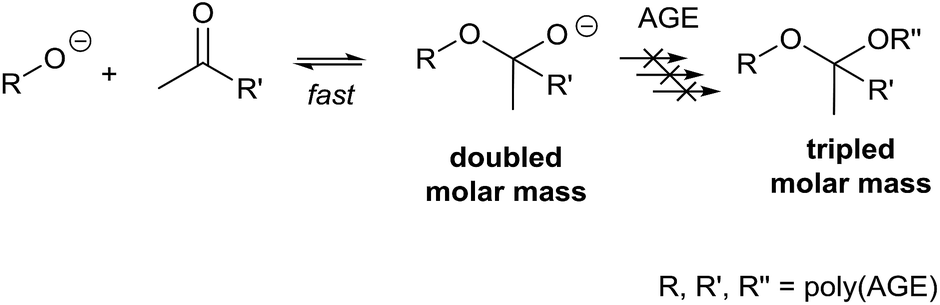 | ||
| Scheme 1 Dimerization of poly(AGE) caused by the nucleophilic attack of the oxyanionic species at initially formed ketone-functional poly(AGE). The polymeric dimer is not assumed to further polymerize due to the fast exchange reaction. | ||
For a detailed investigation, commercially available KOtBu (1 M in THF) was used as initiator. The polymerization was conducted at 45 °C as isomerization reactions are suppressed at this temperature. After adding the total monomer volume in a step-wise manner, the reactions were additionally stirred for 20–24 h at 45 °C to fully polymerize all monomer. Experiments were conducted with AGE and EEGE as monomers with feed rates between ![[V with combining dot above]](https://www.rsc.org/images/entities/i_char_0056_0307.gif) = 50–5000 μL h−1. A syringe pump was used to control the feed rate. Briefly, in a glovebox 100 μL of the initiator was placed into a flask which was sealed with a rubber septum. A syringe equipped with the monomer was punctured through the septum. The syringe/flask-assembly was wrapped with Parafilm® from the neck of the flask to the connection-side of the needle with the syringe to maintain air-tightness. The syringe-flask construct was placed into the syringe-pump and the flask heated in an oil bath. For AGE-experiments 570 μL ([M]:[I] = 50:1) and for EEGE-experiments 731 μL ([M]:[I] = 50:1) of the monomer were added via the programmed syringe pump. After complete addition of the monomer, the flasks were stirred at 45 °C either in an oil bath or an incubator. The samples were analysed by 1H-NMR spectroscopy and SEC measurements. All experiments were performed in triplicate to determine standard deviations of the final number-average molar mass (
= 50–5000 μL h−1. A syringe pump was used to control the feed rate. Briefly, in a glovebox 100 μL of the initiator was placed into a flask which was sealed with a rubber septum. A syringe equipped with the monomer was punctured through the septum. The syringe/flask-assembly was wrapped with Parafilm® from the neck of the flask to the connection-side of the needle with the syringe to maintain air-tightness. The syringe-flask construct was placed into the syringe-pump and the flask heated in an oil bath. For AGE-experiments 570 μL ([M]:[I] = 50:1) and for EEGE-experiments 731 μL ([M]:[I] = 50:1) of the monomer were added via the programmed syringe pump. After complete addition of the monomer, the flasks were stirred at 45 °C either in an oil bath or an incubator. The samples were analysed by 1H-NMR spectroscopy and SEC measurements. All experiments were performed in triplicate to determine standard deviations of the final number-average molar mass (![[M with combining macron]](https://www.rsc.org/images/entities/i_char_004d_0304.gif) n) and dispersity (Ð). 1H-NMR experiments were checked for conversion but in no case residual monomer was detectable.
n) and dispersity (Ð). 1H-NMR experiments were checked for conversion but in no case residual monomer was detectable.
Poly(AGE) was analysed using SEC with DMF (+1 g L−1 LiBr) as eluent and PEG as calibration standard. All elugrams were evaluated in the same run time region (16–22 min, total run time 30 min) to gain comparability. For poly(AGE) n and Ð are shown in Fig. 1. n was between 3800–4400 g mol−1 ( = 50–2000 μL h−1) corresponding to DPs of 33–38, whereas 1H-NMR analyses gave DPs of 47–56 using the terminal tert-butyl group as internal reference. The results were in accordance with the theoretical DP of 50. The differences between the DPs obtained by 1H-NMR spectroscopy and SEC measurements were assigned to a different coiling behaviour of poly(AGE) compared to PEG in DMF.
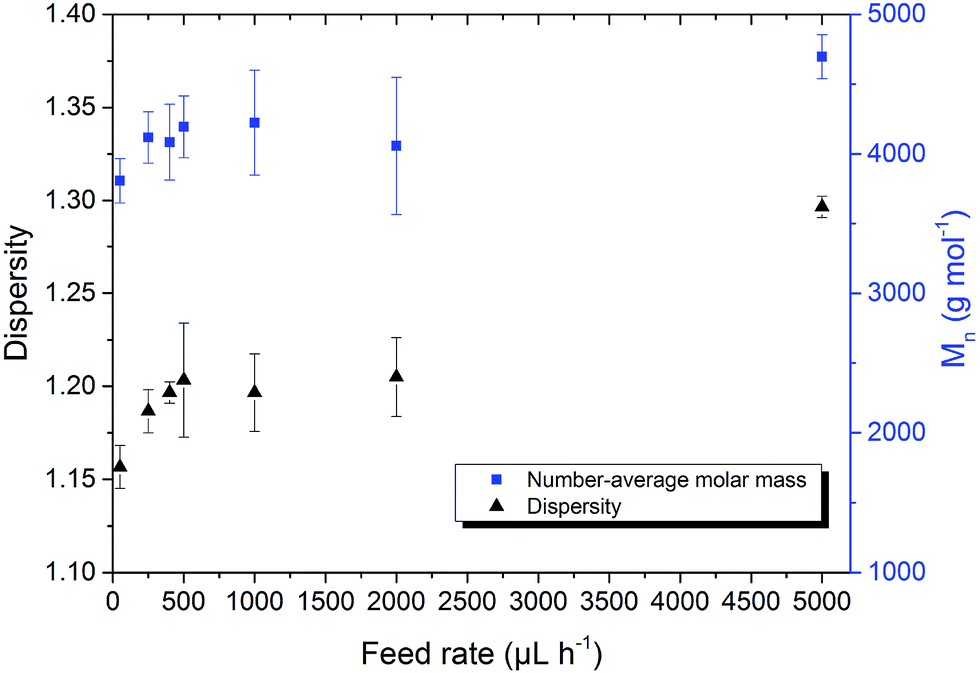 | ||
| Fig. 1 Dispersity and Mn evolution of poly(AGE) depending on the monomer feed rate. Polymerizations were conducted at 45 °C with THF as solvent and KOtBu as initiator. | ||
With an increasing feed rate a slight but steady increase in n was observed. This can be explained by the also observed steady increase of Ð from 1.16 ± 0.01 to 1.30 ± 0.01. Increasing side-reactions lead to a higher fraction of coupled polymers. The resulting polymers with twice the molar-mass of the main polymer fraction led to a shift of n towards higher values. Dispersity Ð evolved from 1.16 ± 0.01 to 1.20 ± 0.01 if the feed rate was increased from 50 μL h−1 to 500 μL h−1, respectively. Between 500 μL h−1 and 2000 μL h−1 no significant dispersity alteration was observed. An increase of the feed rate from 2000 μL h−1 to 5000 μL h−1 yielded polymers with dispersities of 1.21 ± 0.02 and 1.30 ± 0.01, respectively. Qualitatively, SEC elugrams further indicate the hypothesized coupling. The signal of the polymer fraction with twice the molar-mass rose with increased feed rates and bimodal molar-mass distributions were obtained (Fig. 2).
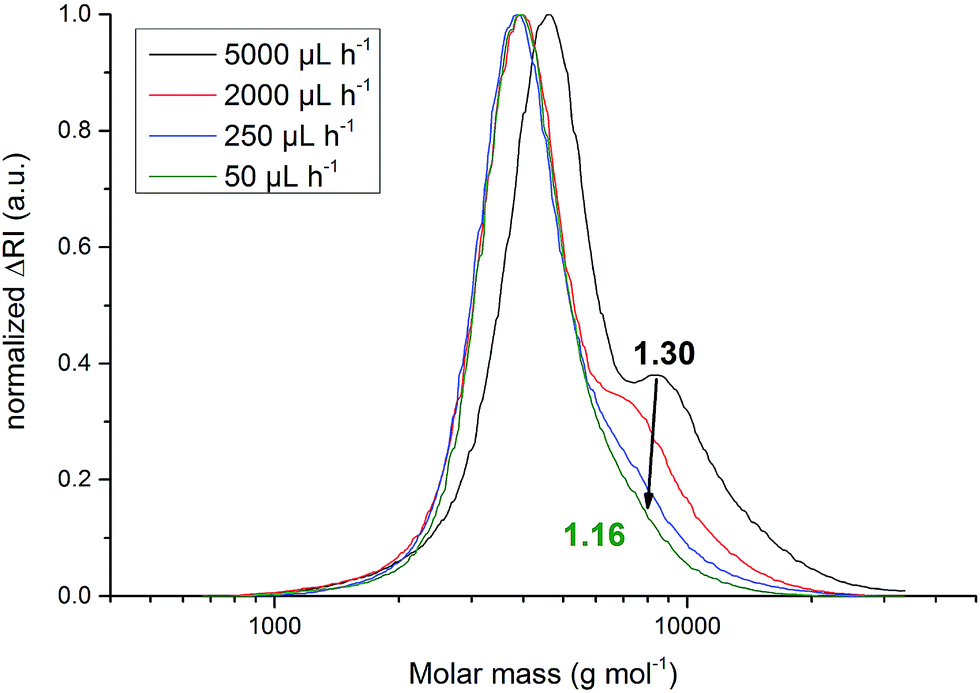 | ||
| Fig. 2 Molar-mass distribution of poly(AGE). Decreasing the feed rate of the monomer from = 5000 to 50 μL h−1 reduces the dispersity from 1.30 to 1.16. | ||
Poly(EEGE) was also synthesized with the same [M]:[I] ratio of 50:1 via the slow monomer addition route. With this, the influence of the monomer on the polymerization was investigated. In general, it was observed that dispersities of poly(EEGE) are lower than dispersities of poly(AGE). The polymer fraction with twice the molar-mass is also present, but less pronounced than for poly(AGE) (see ESI†). For EEGE, feed-rate reduction also lowers the dispersity (Fig. 3). Dispersities were between 1.11 ± 0.01 and 1.16 ± 0.01 for feed rates of 100 μL h−1 and 5000 μL h−1, respectively. Especially below a threshold of 500 μL h−1 dispersity reductions were observed for both monomers (Fig. 1 and 3). We assume that the balance between chain propagation and monomer feed is responsible for this drop in dispersity. If the monomer supply (feed rate) is too high, monomer concentration will rise with time. With a higher monomer content the number of above mentioned side-reaction is more likely to occur. Below the threshold, it is assumed that monomer consumption is higher than the monomer supply and [M] is kept low. With this the deprotonation reaction can be reduced.
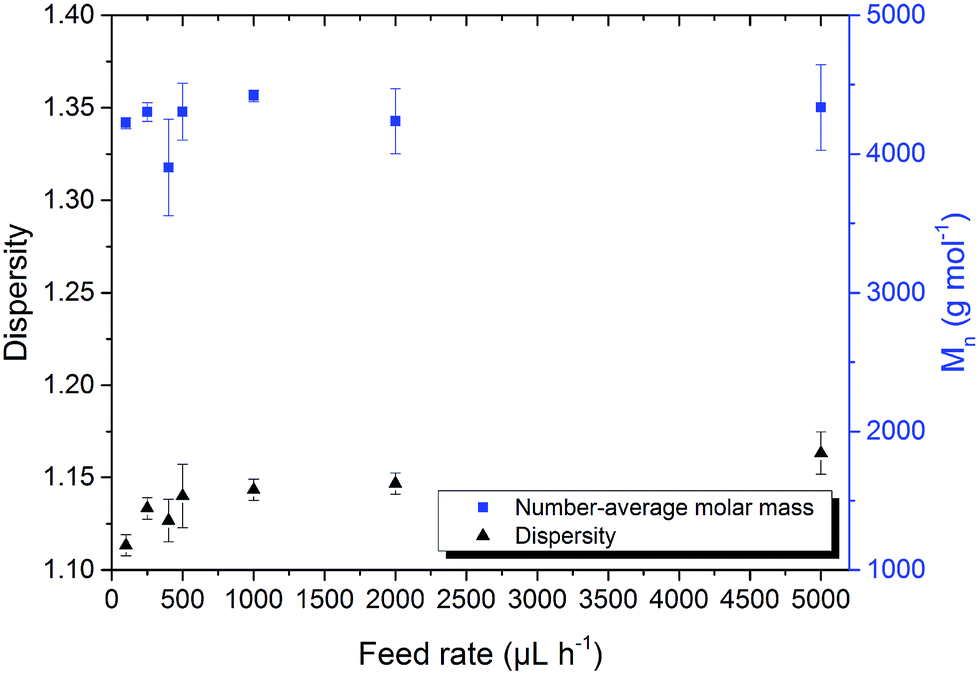 | ||
| Fig. 3 Dispersity and Mn evolution of poly(EEGE) depending on the monomer feed rate. Polymerizations were conducted at 45 °C with THF as solvent and KOtBu as initiator. | ||
First-order kinetics plots were investigated for the polymerizations of AGE and EEGE with an “infinite feed rate” and an initial [M]:[I] ratio of 50:1. The term “infinite feed rate” is referred to experiments without a syringe-pump. Here all monomer is present in the reaction mixture from the beginning. The data plots show a small induction period for the polymerizations of both monomers (Fig. S4†). These induction periods were ascribed to K-alkoxide clusters that can form in THF and are present at the beginning of the polymerization.20,21 These clusters disintegrate with ongoing polymerization due to the increasing steric demand of the polymer chains. After the induction period, linear regions in the plots indicate a living polymerization. For AGE, the linear region was between 43% and 89% conversion, whereas it was between 36% and 92% conversion for EEGE. The slopes obtained from the linear region of the plot were used to determine the propagation rates. AGE polymerizes slightly faster than EEGE with propagation rates of kAGE = 2.4 × 10−3 L mol−1 s−1 and kEEGE = 2.2 × 10−3 L mol−1 s−1, respectively. Earlier investigations by Erberich et al. show a slower polymerization of AGE compared to EEGE.3 This was assigned to the modified polymerization conditions used. The kinetic data indicate that the dispersity drop in AGE polymerization should be achievable at higher feed rates as the propagation rate is slightly higher and the monomer faster consumed. First-order kinetics plots for the slow monomer-addition experiment were not performed. The condition of a constant concentration of the active species cannot be fulfilled. The number of active species is assumed to be constant but the reaction volume and the monomer content continuously change with time. With this, the first order plot cannot directly be applied and a linear relation in the −ln(Mt/M0)–time plot is not expected.
Regarding the feed rate and intended DPs of 50, the total monomer volume has to be considered as well. For above mentioned experiments the total monomer volumes were 570 μL and 731 μL for AGE and EEGE, respectively. With a feed rate of 500 μL h−1, all AGE was injected after 66 min, whereas the addition of all EEGE requires 88 min. This means that polymerization of AGE is faster and additionally monomer injection requires less time.
In conclusion, it was shown that dispersity reduction of poly(glycidyl ether)s can be obtained using a slow monomer addition technique. Feed rate adjustment keeps the monomer concentration constantly low and monomer deprotonation reactions are significantly reduced. This side-reaction commonly leads to bimodal SEC elugrams with a polymer fraction having twice the molar mass of the main polymer. In general, lower dispersities were obtained for poly(EEGE) compared to poly(AGE) and the effect of slow monomer addition was more pronounced for poly(AGE). For a DP of 50, dispersities of poly(AGE) could be reduced from 1.30 ± 0.01 to 1.16 ± 0.01, whereas for poly(EEGE) dispersities were reduced from 1.16 ± 0.01 to 1.11 ± 0.01. By lowering the feed rate SEC elugrams showed qualitatively the change from bimodal to monomodal elugrams. In the future, poly(AGE) and poly(EEGE) are attempted having molar masses higher than 20000 g mol−1. This shall further evaluate the scope of this technical dispersity control. Additionally, further polymerization parameters will be investigated and the hypothesis evaluated for other glycidyl-ether monomers.
Acknowledgements
This work was financially supported by the European research council (ERC consolidator grant Design2Heal, contract no. 617989). The authors thank Paul Dalton and Jörg Kuhlmann for proof reading the manuscript.Notes and references
- B. F. Lee, M. J. Kade, J. A. Chute, N. Gupta, L. M. Campos, G. H. Fredrickson, E. J. Kramer, N. A. Lynd and C. J. Hawker, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4498–4504 CrossRef CAS PubMed.
- M. Backes, L. Messager, A. Mourran, H. Keul and M. Moeller, Macromolecules, 2010, 43, 3238–3248 CrossRef CAS.
- M. Erberich, H. Keul and M. Möller, Macromolecules, 2007, 40, 3070–3079 CrossRef CAS.
- B. F. Lee, M. Wolffs, K. T. Delaney, J. K. Sprafke, F. A. Leibfarth, C. J. Hawker and N. A. Lynd, Macromolecules, 2012, 45, 3722–3731 CrossRef CAS PubMed.
- Y. Koyama, M. Umehara, A. Mizuno, M. Itaba, T. Yasukouchi, K. Natsume, A. Suginaka and K. Watanabe, Bioconjugate Chem., 1996, 7, 298–301 CrossRef CAS PubMed.
- B. Obermeier and H. Frey, Bioconjugate Chem., 2011, 22, 436–444 CrossRef CAS PubMed.
- M. Kuhlmann, O. Reimann, C. P. R. Hackenberger and J. Groll, Macromol. Rapid Commun., 2015, 36, 472–476 CrossRef CAS PubMed.
- M. Hans, H. Keul and M. Moeller, Polymer, 2009, 50, 1103–1108 CrossRef CAS PubMed.
- S. Liu, F. Zhang, Y. Zhang and J. Xu, Chin. J. Chem., 2013, 31, 1315–1320 CrossRef CAS PubMed.
- M. Gervais, A.-L. Brocas, G. Cendejas, A. Deffieux and S. Carlotti, Macromolecules, 2010, 43, 1778–1784 CrossRef CAS.
- W. Kwon, Y. Rho, K. Kamoshida, K. H. Kwon, Y. C. Jeong, J. Kim, H. Misaka, T. J. Shin, J. Kim, K.-W. Kim, K. S. Jin, T. Chang, H. Kim, T. Satoh, T. Kakuchi and M. Ree, Adv. Funct. Mater., 2012, 22, 5194–5208 CrossRef CAS PubMed.
- A. Stolarzewicz, Makromol. Chem., 1986, 187, 745–752 CrossRef CAS PubMed.
- F. Wurm, J. Nieberle and H. Frey, Macromolecules, 2008, 41, 1184–1188 CrossRef CAS.
- P. Dimitrov, S. Rangelov, A. Dworak and C. B. Tsvetanov, Macromolecules, 2004, 37, 1000–1008 CrossRef CAS.
- A. Dworak, G. Baran, B. Trzebicka and W. Wałach, React. Funct. Polym., 1999, 42, 31–36 CrossRef CAS.
- M. Iijima, Y. Nagasaki, M. Kato and K. Kataoka, Polymer, 1997, 38, 1197–1202 CrossRef.
- B. Obermeier, F. Wurm and H. Frey, Macromolecules, 2010, 43, 2244–2251 CrossRef CAS.
- F. Wurm, H. Schüle and H. Frey, Macromolecules, 2008, 41, 9602–9611 CrossRef CAS.
- B. Schulte, A. Walther, H. Keul and M. Möller, Macromolecules, 2014, 47, 1633–1645 CrossRef CAS.
- P. Schmidt, L. Lochmann and B. Schneider, J. Mol. Struct., 1971, 9, 403–411 CrossRef CAS.
- M. H. Chisholm, S. R. Drake, A. A. Naiini and W. E. Streib, Polyhedron, 1991, 10, 337–345 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental section of synthesis, setup 1H-NMR and elugrams. See DOI: 10.1039/c5ra08067c |
| This journal is © The Royal Society of Chemistry 2015 |