
A dual-mode luminescent probe composed of co-assembled down-conversion CdTe and up-conversion NaYF4:Yb,Tm(Er) nanoparticles†
Abstract
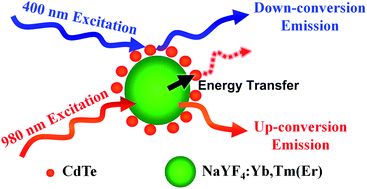
Multiplex luminescent probes with good water-dispersibility are basic materials in current biological research as labels and sensors. In this work, we demonstrate a facile approach to fabricate water-dispersible dual-mode luminescent probes by co-assembling down-conversion CdTe nanoparticles (NPs) and up-conversion NaYF4:Yb,Tm(Er) NPs. 3-Mercaptopropionic acid (MPA)-capped CdTe NPs are water soluble, while oleic acid (OA)-capped NaYF4:Yb,Tm(Er) NPs are dispersed in chloroform. There also exists an excess of Cd–MPA complex in the CdTe aqueous solution. After mixing CdTe and NaYF4:Yb,Tm(Er), the MPA on CdTe NPs and Cd–MPA complexes is capable of partially replacing the OA on NaYF4:Yb,Tm(Er) NPs, thus transferring NaYF4:Yb,Tm(Er) NPs to water and forming CdTe–NaYF4:Yb,Tm(Er) hybrid NPs. The hybrid NPs combine the down-conversion emission of CdTe and the up-conversion emission of NaYF4:Yb,Tm(Er), which can be excited both by 400 and 980 nm irradiation and exhibit dual-mode emission. Both Förster resonance energy transfer from NaYF4:Yb,Tm(Er) to CdTe and photon re-absorption are observed as the hybrid NPs are excited by 980 nm irradiation.
 Please wait while we load your content...
Please wait while we load your content...

