
Evolution of boron clusters in iron tetraborides under high pressure: semiconducting and ferromagnetic superhard materials
Abstract
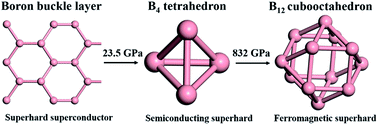
We investigated the high-pressure structures and properties of iron tetraborides (FeB4) using a combination of an ab initio high-throughput search and a particle-swarm optimization algorithm for crystal structure prediction. We found that, under compression, the boron sublattice in FeB4 from the buckled boron layer first polymerizes into B4 tetrahedral clusters and then forms cubo-octahedral B12 clusters. At 55 GPa, the orthorhombic crystal structure with a Pnnm space group (58-FeB4) transforms into a tetragonal I41/acd structure (142-FeB4), which is stable within a wide pressure range up to 695 GPa. Then, a cubic Im![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m phase (229-FeB4) emerges at higher pressures up to at least 1 TPa. The computed Vicker's hardnesses of 58-, 142-, and 229-FeB4 are 61.58, 47.44, and 50.87 GPa, respectively. All of them can be considered as superhard materials. Compared to the previously reported 58-FeB4 as a superhard superconductor, the B4 tetrahedral cluster-based 142-FeB4 is a superhard semiconductor with an indirect band gap of 1.34 eV. The pressure-induced metal-to-semiconductor transition can be related to a unique Fe–B–B three-center covalent bond. Moreover, 229-FeB4, which is composed of cubo-octahedral B12 clusters, is ferromagnetic with a magnetic moment of 0.929μB per Fe atom at ambient pressure. The magnetic moment will decrease rapidly with increasing pressure and be completely quenched as pressure exceeds 40 GPa. The pressure-induced evolution of boron cluster units not only adds new features to boron chemistry, but also gives rise to novel superhard semiconductors or ferromagnetic materials. Moreover, our results may inspire further experimental and theoretical interest in designing new materials using clusters as pseudo-atoms with expected properties.
m phase (229-FeB4) emerges at higher pressures up to at least 1 TPa. The computed Vicker's hardnesses of 58-, 142-, and 229-FeB4 are 61.58, 47.44, and 50.87 GPa, respectively. All of them can be considered as superhard materials. Compared to the previously reported 58-FeB4 as a superhard superconductor, the B4 tetrahedral cluster-based 142-FeB4 is a superhard semiconductor with an indirect band gap of 1.34 eV. The pressure-induced metal-to-semiconductor transition can be related to a unique Fe–B–B three-center covalent bond. Moreover, 229-FeB4, which is composed of cubo-octahedral B12 clusters, is ferromagnetic with a magnetic moment of 0.929μB per Fe atom at ambient pressure. The magnetic moment will decrease rapidly with increasing pressure and be completely quenched as pressure exceeds 40 GPa. The pressure-induced evolution of boron cluster units not only adds new features to boron chemistry, but also gives rise to novel superhard semiconductors or ferromagnetic materials. Moreover, our results may inspire further experimental and theoretical interest in designing new materials using clusters as pseudo-atoms with expected properties.
 Please wait while we load your content...
Please wait while we load your content...

