
Electronic and charge transport properties of dimers of dithienothiophenes: effect of structural symmetry and linking mode
Ping Li,
Yahui Cui,
Chongping Song and
Houyu Zhang*
State Key Laboratory of Supramolecular Structure and Materials, Institute of Theoretical Chemistry, Jilin University, Changchun 130012, P. R. China. E-mail: houyuzhang@jlu.edu.cn; Tel: +86-431-85168492
First published on 29th May 2015
Abstract
The electronic structures and charge transport properties of a series of dimers of dithienothiophenes are investigated by means of quantum chemical calculations. To gain a better understanding of the effects of the structural symmetry and linking mode on the dimers, the geometrical structures, molecular reorganization energies upon gaining or losing electrons, molecular ionization potentials (IPs) and electron affinities (EAs), molecular aromaticities, frontier molecular orbitals, as well as charge mobilities are analyzed in detail to determine the structure–property relationships for the investigated dimers of dithienothiophenes. The calculated results show that the vinylene-linked dimers have advantages over the directly single-bond linked dimers because of the large extent of π conjugation and thus enhanced π–π stacking interactions in their crystal structures. The molecular symmetry could affect the electron density distributions in the molecules, and further determine the molecular orientations and intermolecular arrangements. High molecular symmetry could facilitate the molecular packing in order, thus enhancing the charge transport. The theoretical characterization of these dimers in combination with experimental results indicate that highly symmetrical vinylene-bridged dimers could be promising candidates for transistor applications, and shed light on the molecular design of high performance materials.
Introduction
Organic semiconductors (OSCs) have drawn continuous attention for decades due to their prospective commercialized applications in electronic devices, such as organic field effect transistors (OFETs),1–7 organic solar cells,8–10 and organic light emitting diodes (OLEDs).11–13 Among the most extensively studied OSCs, oligothiophene-based conjugated materials are important representative systems owing to their chemical versatility, favorable electrical and optical properties.14,15 α-Oligothiophenes have been successfully applied as active components in OLEDs and OFETs, but low conjugation because of the torsion of single bonds or S-syn defects limit their optical and charge-transport performances in practical devices.16 However fused oligothiophenes (thienoacenes) possess the advantages of extended conjugation and rigid planarity, thus leading to improved charge transporting properties and chemical stability.17–19 The sulfur atoms in thienoacenes have high polarizability and thus facilitate electron-donating properties. Multiple short intermolecular S⋯S contacts originating from the sulfur atoms at the molecular periphery can make the molecules densely packed in the solid state, resulting in the enhanced charge transport properties.The structures of thienoacenes rely on selective connections and annelations of thiophene building blocks at the α-, and β-positions, which will lead to quasi-linearly annelated α-thienoacenes and helically annelated β-thienoacenes.20 The oligothienoacenes of dithieno[3,2-b:2′,3′-d]-thiophene and dithieno[2,3-b:3′,2′-d]-thiophene (hereafter denote as α- and β-trithiophene) are regarded as effective molecular building units with quite different LUMO character and HOMO–LUMO gaps (as shown in Fig. 1a), and can be used to design versatile OSCs. The dimerizations of α- and β-trithiophenes linked by a single bond (1–3) and vinylene bridge (4–6) were constructed as new candidates for OFET materials (Fig. 1b).21 The dimer of α-trithiophene 1 has been demonstrated to be effective as the active layer in an FET device and shows hole mobility of up to 0.05 cm2 V−1 s−1.22,23 The mobility of the dimer of β-trithiophene 2 is 0.005 cm2 V−1 s−1 at room temperature, while the device performance of vinylene-bridged dimer of β-trithiophene 5 is apparently much better than that of 2, exhibiting excellent OFET performance with mobility as high as 0.89 cm2 V−1 s−1.24 The answer to why a subtle structural change leads to a significant effect on the charge transport properties should be revealed at a microscopic level and a detailed understanding of the structure–property relationship should be established for these OSCs.
 | ||
| Fig. 1 (a) The chemical structures of α- and β-trithiophenes with their frontier orbitals and HOMO–LUMO gaps and (b) the dimer structures investigated in this work. The letters denote symmetry-unique rings in these molecules. | ||
In the present work, we focus on the effects of structural symmetry and linking mode on the electronic structures of the dimers of dithienoacenes. Compounds 1, 2, 4 and 5 are highly symmetrical in structure, while the cross-linked compounds 3 and 6 are asymmetrical. How and to what extent the structural symmetry affects the intermolecular interactions of these thienoacenes needs to be elucidated. The question of why the different linking modes of either a direct single bond or a vinylene bridge could result in distinct electronic properties still needs to be answered. The molecular geometries, the molecular frontier orbitals, molecular IPs and EAs, and reorganization energies will be definitely influenced by the structural symmetry and linking mode in the dimerization of dithienoacenes. Additionally, the α- and β-trithiophenes have different π-conjugation and exhibit distinct aromaticity originating from the location of the sulfur atoms in the molecular backbone. The aromaticity in the dimer structure will subtly change in comparison to that of the monomer counterpart. All these electronic properties need to be investigated to further understand their relationship with structural changes at a microscopic level.
Theoretical calculation is becoming a powerful tool to study the electronic properties of OSCs and predict their charge transport properties.25–32 At a high-temperature regime and in the presence of structural disorders, charge carriers are localized over a single molecule,33,34 which makes the bandlike mechanism fail in describing the transport behavior in many OSCs.35,36 Thus, a thermally activated hopping and diffusion model can be employed to simulate the charge carrier motion.37–39 Based on quantum-chemical calculations, we make comparative studies of the electronic and charge transport of properties of the dimers of α- and β-trithiophene. By means of density functional theory (DFT) calculations, we aim to establish structure–property relationships of thienoacene-based materials and shed light on the fundamental research on designing high charge mobility materials.
Theoretical and computational method
The molecular geometries of neutral and charged states are optimized at the DFT level using the B3LYP hybrid functional40,41 and 6-31G(d, p) basis set, as implemented in the Gaussian 09 package.42 Harmonic vibrational frequencies are calculated at the same level of theory on the basis of the resulting optimized geometries. The total density of state (DOS) and projected density of state (PDOS) for sulfur atom and phenyl (or thienyl) group are obtained with the GaussSum 2.25 program.43To study the charge transport properties of the dimers of dithienothiophenes at room temperature, the incoherent hopping mechanism is adopted to describe the sequential charge jumps between adjacent molecules. For each charge hopping event, the self-exchange charge transfer rate can be expressed by the Marcus–Hush equation44,45 in terms of reorganization energy λ and electronic coupling Vab between neighbouring molecules a and b:46
 | (1) |
 | (2) |
| Vab = 〈Ψ0,ai|F0|Ψ0,bi〉 | (3) |
The choice of DFT functional is important for an accurate description of the ground-state electronic properties and electronic couplings between adjacent molecules. In comparison to pure DFT functionals, hybrid functionals can give a better estimate of the HOMO–LUMO energy gap because of the incorporation of a fraction of non-local Hartree–Fock (HF) exchange. While long-range corrected functionals are better than hybrid functionals in describing the charge-transfer excited state in push–pull molecules.55,56 In this work, the electronic coupling is calculated from the overlap of the ground-state frontier orbitals of adjacent molecules. Such calculations do not involve the excited charge transfer. So hybrid functionals such as B3LYP,57,58 M062X,59 and MPWB1K31 and combined exchange–correlation functional PW91PW91 29,33,60 are used to calculate the electronic couplings in the literature. Hence the hybrid functional B3LYP is employed to calculate the ground-state electronic structures and electronic couplings in the dimer structures.
The analyses of the local aromaticity in all compounds are performed by means of nucleus independent chemical shifts (NICS(1)), and the harmonic oscillator model of aromaticity (HOMA) index. The calculated NICS(1) and HOMA values provide a relative comparison of aromaticity among all of the compounds. In the NICS(1) procedure suggested by Schleyer et al.,61 the absolute magnetic shielding is computed at 1 Å above and 1 Å below the center of the ring (for the heterocyclic ring in this work, we define the center as the ring bonding critical point). The HOMA index is calculated as
 | (4) |
Results and discussion
Molecular geometry and reorganization energy
The optimized structures of the investigated molecules in the neutral state for the gas phase are shown in Fig. 2. Though α- and β-trithiophene exhibit a planar and rigid skeleton, the dimers linked by a single bond render twisted structures with dihedral angles about 20.5°, 29.0° and 24.2° for 1, 2 and 3, respectively. The dihedral angle between the two trithiophenes in 1 is similar to the value between two thiophenes in α-bithiophene (22°). Compounds 2 and 3 show more distorted structures, while the dimers bridged by vinylene show planar structures. The dimers 1 and 2 have C2 symmetry, while 4 and 5 have C2h symmetry due to the planarity. The cross-linked dimers 3 and 6 have no symmetry in their structures. When a molecule gains or loses charges, it will relax its molecular geometry for a new charge distribution. All the cationic and anionic states for compounds 1–6 are almost planar structures. The changes of bond lengths upon oxidation (losing an electron from the neutral to the cationic state) and reduction (gaining an electron from the neutral to the anionic state) of 1, 2, 3 and 6 are presented in Fig. 3. Because of the extended π-system, changes of bond lengths upon oxidation and reduction are found to occur over the entire molecule, and particularly more pronounced in the thiophene rings at the linkage. For the asymmetric cross-linked 3 and 6, the bond length changes in the α-trithiophene are more notable than those in β-trithiophene. The bond length changes of 3 are a bit larger than those of 6 because of the distorted geometrical relaxation from non-planarity to planarity. The bond length changes upon oxidation and reduction are comparable for the selected compounds. Gaining or losing charges causes changes in geometrical structure, which is a reflection of the reorganization energy. | ||
| Fig. 2 Optimized structures in the neutral state of the investigated molecules. | ||
 | ||
| Fig. 3 Bond-length changes (in Å) upon oxidation and reduction for compounds 1, 2, 3 and 6. The bond indices are labelled on the molecular structures. | ||
From eqn (1), we know that the charge hopping rate benefits from a smaller reorganization energy. The reorganization energies of the investigated compounds are collected in Table 1. Compound 2 has the largest reorganization energies for both hole and electron, which is in agreement with the larger bond-length changes in Fig. 3. The reorganization energies of the dimers of α-trithiophenes are smaller than those of the counterpart dimers of β-trithiophenes, while the reorganization energies of the cross-linked dimers (3 and 6) are in between the dimers of α- and β-trithiophene. The reorganization energies are to a large extent dependent on the intrinsic properties of α- and β-fused trithiophenes. The reorganization energies for both electron and hole of vinylene-bridged dimers (4, 5, and 6) are smaller than their corresponding counterpart (1, 2, and 3) of single-bond linked dimers. The vinylene bridge can significantly decrease the steric hindrance between the trithiophene units at the linkage and extend π conjugation, resulting in more rigid and planar structures with higher molecular symmetries. All the molecules have relatively smaller electron reorganization energies because of the existence of high polarizability of the sulfur atom. As for the introduction of the substituted benzene and thiophene units at the longitudinal ends of 5, the reorganization energies are reduced, especially for the electron reorganization energy in 5b.
| Compd | λh | λe | AIP | VIP | AEA | VEA |
|---|---|---|---|---|---|---|
| 1 | 0.338 (0.332) | 0.298 (0.295) | 6.42 (6.23) | 6.60 (6.41) | −1.08 (−0.77) | −0.91 (−0.60) |
| 2 | 0.381 (0.373) | 0.358 (0.380) | 6.67 (6.50) | 6.89 (6.71) | −0.58 (−0.20) | −0.36 (0.02) |
| 3 | 0.359 (0.350) | 0.326 (0.326) | 6.53 (6.35) | 6.74 (6.55) | −0.86 (−0.53) | −0.67 (−0.34) |
| 4 | 0.292 (0.298) | 0.245 (0.255) | 6.21 (6.02) | 6.36 (6.17) | −1.32 (−1.01) | −1.19 (−0.89) |
| 5 | 0.308 (0.302) | 0.274 (0.293) | 6.41 (6.24) | 6.56 (6.39) | −0.93 (−0.60) | −0.80 (−0.46) |
| 5a | 0.307 (0.311) | 0.255 (0.252) | 6.33 (6.15) | 6.48 (6.30) | −1.03 (−0.72) | −0.91 (−0.59) |
| 5b | 0.290 (0.286) | 0.209 (0.209) | 6.36 (6.18) | 6.50 (6.31) | −1.08 (−0.78) | −0.98 (−0.67) |
| 6 | 0.298 (0.304) | 0.259 (0.272) | 6.31 (6.12) | 6.46 (6.27) | −1.15 (−0.83) | −1.02 (−0.70) |
Molecular IP and EA
The efficient injection of holes and electrons is important for the rational design of optimized electronic devices. The molecular IP and EA are key parameters to estimate the energy barrier for the injection of holes and electrons into a molecule and provide useful information regarding ambient stability.63 The calculated IPs and EAs of compounds for both adiabatic (optimized structure for both the neutral and charged molecules) and vertical (at the geometry of the neutral molecule) values are presented in Table 1. Since the IPs and EAs are very dependent on the diffuse function of the basis set,64 the calculated results are performed with the large basis set of 6-31++G(d, p) in comparison with 6-31G(d, p). The energy difference between the adiabatic and vertical value could reflect the extent of the structural relaxation upon charge injection.65 The calculated adiabatic and vertical IPs are larger than the adiabatic experimental IP for a stable p-type OFET material sexthiophene (5.80 eV),66 indicating that all the compounds are more stable and exhibit antioxidative ability in air. Upon going from the single-bond linked dimers to the vinylene-bridged dimers, the adiabatic IPs decrease slightly by 0.2–0.4 eV, but the compounds are still stable and can favorably reduce the hole-injection barrier for the commonly used electrode (Au work function: ∼5.1 eV).In addition to the decrease in IPs, the vinylene bridge also makes the EAs of the dimers more exothermic. A negative value of EA indicates exothermicity for the reduction of a molecule, for example, 5 is 0.35 eV more exothermic than 2. The vinylene-bridged dimers have small EA values ranged from −0.93 eV to −1.32 eV (see Table 1 for the adiabatic value of EAs). The calculated EA values are not matchable to the workfunction of the commonly used metallic electrodes (∼3 eV) and are less stable upon reduction. We can see that vinylene-bridged dimers have more negative EAs, which are of great benefit to lowering the energy barrier for electron injection and improving the stability of their anions by preventing chemical reactions with water and oxygen.
From the redox stability point of view, the single-bond linked dimers are more stable in their cationic states and less stable in their anion states than their corresponding counterpart of vinylene-bridged dimers. In comparison to α-trithiophene dimers, β-trithiophene dimers have larger IPs and EAs. The cross-linked dimers have IPs and EAs in between those of dimers of α- and β-trithiophene. This trend is in good agreement with that of reorganization energy. It is noticeable that the introduction of a phenyl or thienyl group at the longitudinal ends of 5 decrease both IPs and EAs, which are further favorable for improving hole or electron injection.
Structure and aromaticity
Aromaticity is the property of a planar, cyclic, conjugated molecule in which cyclic electron delocalization results in enhanced stability, bond length equalization, and special magnetic, chemical and physical properties.67,68 The α- and β-trithiophenes are aromatic because the molecules are cyclic and planar structures, and follow Huckel’s rule, having 4n + 2 electrons in the delocalized π-orbitals. The aromatic properties will slightly change when two trithiophene subunits are linked through a single bond or a vinylene bridge. Considering the fact that the molecular geometry determined by the extent of π-conjugation is an important factor to determine the electronic structures of dimers, we evaluate the aromaticity of the heterocyclic rings of the dimers to understand the molecular structure–property relationship.The calculated NICS(1) and HOMA values are collected in Table 2. For the building block α- and β-trithiophenes, the absolute values of NICS(1) and HOMA of central rings (b) are smaller than those of the periphery rings (a), as indicated in Fig. 1b. The average absolute values of NICS(1) and HOMA for all aromatic rings in α-trithiophenes are larger than that in β-trithiophenes, which indicates that electrons are more delocalized in α-trithiophenes. α-Trithiophene has a larger extent of π-conjugation than β-trithiophene, which is in agreement with the smaller HOMO–LUMO gap in α-trithiophene (as shown in Fig. 1a). The aromaticity differences between α- and β-trithiophene originate from the location of sulfur atoms in the molecular backbone, as can be seen from their LUMO character in Fig. 1a. While in the dimer structures, the absolute values of NICS(1) and HOMA of the rings at the linkage dramatically decrease, resulting in an average aromaticity decrease in both single-bond and vinylene-bridge linked dimers. The average aromaticity in the vinylene-bridged dimers is smaller than that of the single-bonded dimers. In a previous report, Chen et al. studied the relationship between aromaticity and conductance of a single-molecule junction and verified that the conductance correlates negatively with the aromaticity.69 So the decrease of the aromaticity of the compounds might favour charge hopping between the heterocyclic rings.
| Compd | Ring | NICS(1) | NICS(1)a | HOMA | HOMAa |
|---|---|---|---|---|---|
| a The average value for all heterocyclic aromatic rings. | |||||
| α | a | −8.624 | −8.204 | 0.726 | 0.711 |
| b | −7.365 | 0.681 | |||
| β | a | −8.721 | −8.123 | 0.691 | 0.669 |
| b | −6.925 | 0.624 | |||
| 1 | a | −8.678 | −7.718 | 0.729 | 0.705 |
| b | −7.211 | 0.690 | |||
| c | −7.418 | 0.696 | |||
| 2 | a | −8.733 | −7.670 | 0.693 | 0.662 |
| b | −6.801 | 0.627 | |||
| c | −7.474 | 0.665 | |||
| 3 | a | −8.524 | −7.774 | 0.729 | 0.683 |
| b | −7.275 | 0.686 | |||
| c | −7.351 | 0.697 | |||
| d | −7.316 | 0.667 | |||
| e | −7.433 | 0.626 | |||
| f | −8.747 | 0.694 | |||
| 4 | a | −8.511 | −7.623 | 0.730 | 0.705 |
| b | −7.233 | 0.691 | |||
| c | −7.126 | 0.694 | |||
| 5 | a | −8.793 | −7.610 | 0.694 | 0.662 |
| b | −6.780 | 0.627 | |||
| c | −7.256 | 0.664 | |||
| 6 | a | −8.530 | −7.612 | 0.731 | 0.686 |
| b | −7.221 | 0.680 | |||
| c | −7.158 | 0.716 | |||
| d | −7.248 | 0.667 | |||
| e | −6.739 | 0.627 | |||
| f | −8.772 | 0.695 | |||
Molecular orbitals and density of state
Frontier orbitals, especially HOMO and LUMO, are closely related to the gain and loss electrons during charge transport. The relative orderings of HOMO and LUMO energies provide a reasonable qualitative indication of the ability of hole and electron injection, respectively. A good p-type conducting material is required to have a relatively high HOMO to reduce the charge injection barrier from the anode. The HOMO and LUMO plots and orbital energy levels of the dimers are displayed in Fig. 4, and their experimental values are listed in Table 3. They show that the calculated HOMO and LUMO energies are in agreement with the experimentally measured values, especially with a change in trend.70 The dimers of α- and β-trithiophene (1 and 2) possess higher HOMOs, lower LUMOs and smaller HOMO–LUMO gaps than their monomers (as shown in Fig. 1a). The vinylene-bridged dimers (4, 5 and 6) also have higher HOMOs, lower LUMOs and smaller HOMO–LUMO gaps with respect to their corresponding single-bond linked dimers (1, 2 and 3). Such observations can be attributed to the extension of π-conjugation. Both raising HOMOs and lowering LUMOs are in favor of charge injections in practical application in devices. In the dimers of α-trithiophene (1 and 4), the HOMO and LUMO spread over the entire molecules. While the HOMO and LUMO are concentrated on the central part of the dimers of β-trithiophenes. Both dimers of α- and β-trithiophene have symmetric HOMO and LUMO distribution in the structures, while the dimers of cross-linked α- and β-trithiophene have asymmetrical distribution of frontier orbitals, with the HOMO and LUMO located more on α-trithiophenes than those on β-trithiophenes. | ||
| Fig. 4 HOMO and LUMO energy levels of all the studied molecules and their electronic density contours investigated by the B3LYP/6-31G(d, p) method. | ||
| Compd | HOMO (eV) | LUMO (eV) | H–L gap | ||
|---|---|---|---|---|---|
| Theo. | Expt.a | Theo. | Theo. | Expt.a | |
| a Data from ref. 20. | |||||
| 1 | −5.13 | −5.43 | −1.86 | 3.27 | 2.80 |
| 2 | −5.39 | −5.49 | −1.37 | 4.02 | 3.15 |
| 3 | −5.24 | −5.46 | −1.67 | 3.57 | 2.93 |
| 4 | −4.97 | −5.33 | −2.08 | 2.89 | 2.65 |
| 5 | −5.14 | −5.39 | −1.77 | 3.37 | 2.91 |
| 5a | −5.14 | −5.41 | −1.78 | 3.36 | 2.90 |
| 5b | −5.17 | −5.42 | −1.81 | 3.36 | 2.89 |
| 6 | −5.04 | −5.36 | −1.95 | 3.10 | 2.76 |
The introduction of phenyl or thienyl groups at the longitudinal ends of 5 will definitely affect the frontier orbitals of the dimer of vinylene-bridged β-trithiophene. To investigate the substituent effect on the composition of orbitals near the HOMO–LUMO gap, we calculate total DOS and PDOS for three β-annulated oligothiophenes, as shown in Fig. 5. We find that the sulfur atoms partially take part in the formation of both HOMOs and LUMOs. From the electronic structure point of view, the sulfur atoms directly involve the charge carrier transport. Both the HOMOs and LUMOs in 5a and 5b are localized in the central part of the compounds. The phenyl and thienyl group are not involved in the formation of HOMOs and LUMOs, but they largely participate in the HOMO−1 and LUMO+1. Because of the existence of dihedral angles between the phenyl group or thienyl group and the central π systems of about 29.2° and 25.2° respectively, the substituted phenyl or thienyl group cannot extend the conjugation of the π system significantly and are only partly involved in the charge transport. From the molecular structure point of view, the phenyl or thienyl group will affect the intermolecular arrangements and orbital interactions, resulting in different electronic couplings for holes and electrons.
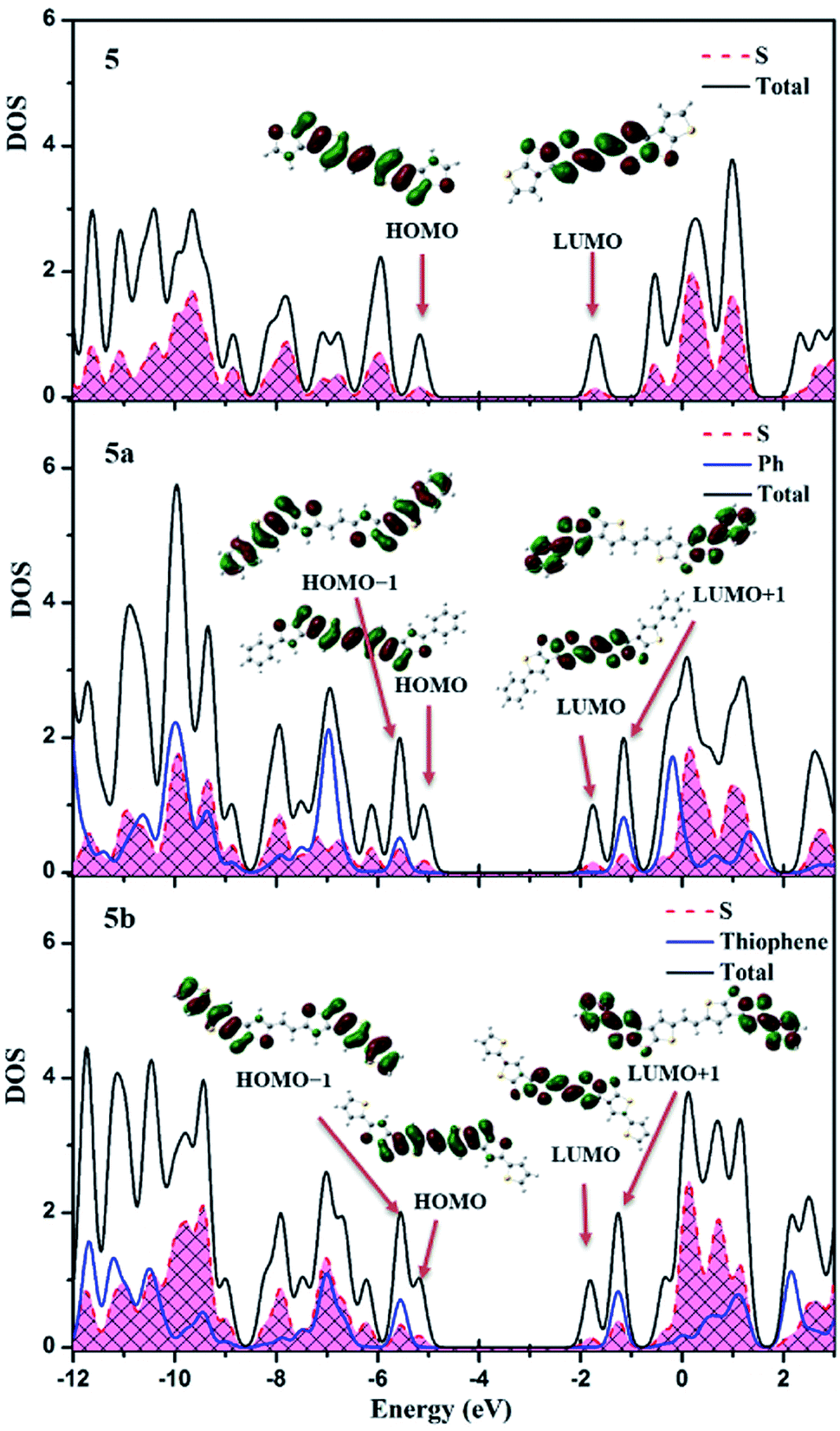 | ||
| Fig. 5 Total DOS and PDOS for sulfur atom, phenyl and thienyl group in the energy window of −12 and 3 eV for 5, 5a and 5b. | ||
Electronic coupling and mobility
The charge transport ability of an organic semiconductor is dependent on not only the electronic properties of the molecule itself but also the molecular packing structures. Besides the intrinsic molecular reorganization energy and ability to gain or lose an electron from a molecule, charge transport is highly sensitive to the relative orientation and molecular stacking character between molecules.71 The structure of the single crystal could provide a consistent platform to study the structural effects. Therefore, the charge transport properties of oligothiophenes are analyzed on the basis of single crystal structures to reach a deeper understanding of the influence of the structural symmetry and linking mode on the nature of the charge transport.The experimental results have proved that the two asymmetric molecules 3 and 6 exhibit low mobilities on the order of 10−4 cm2 V−1 s−1.21 The likely explanation might be the asymmetric structure induced strong disorder in molecular packing in the amorphous films. From the frontier orbital standpoint of view, the electron densities are nonuniformly and asymmetrically distributed in α- and β-trithiophenes, as can be seen from the HOMOs and LUMOs of 3 and 6 in Fig. 4. Such electron density distributions are not in favor of the π-orbital interactions, thus affecting the charge transport between the adjacent molecules. Single crystals are not available for asymmetrical 3 and 6 in the experiment because of structural disorder and the lack of intermolecular π-orbital interactions. However symmetric molecules 1, 2, 4 and 5 are well packed in their crystal structures. It is established that the better the molecular organization in order, the higher the material performance.7 So the molecular symmetry affects the intermolecular interactions, thus determining the molecular packing structures in their solid states.
The available crystal structures of symmetric compounds are shown in Fig. 6, in which molecules in the same layer in the crystal are viewed from the molecular long axis. Choose one molecule as charge donor, and all the surrounding nearest neighbor molecules can be regarded as charge acceptors. The possible intermolecular hopping pathways from the central molecule are also displayed in Fig. 6, and the corresponding intermolecular electronic couplings are collected in Table 4. The crystal structures of the dimers of α-trithiophene 1 and 4 have displaced π–π stacking features with the respective interplanar distances of 3.54 Å and 3.49 Å. The displaced π-stacking configurations and pictorial orbital interactions between HOMOs are displayed in Fig. 7. The π–π stackings in crystals 1 and 4 prefer the slipped configurations because of the existence of strong π-orbital repulsions when molecules are on top of each other. The intermolecular displaced π–π stackings for 1 and 4 are quite different: compound 1 prefers slipping along the molecular short axis about one third of a thiophene ring; while compound 4 is shifted in the direction of the molecular long axis about three fourths of a thiophene ring. The π–π stacking could provide a large overlap of π orbital, which is in favor of charge transfer. The electronic couplings for a hole for intermolecular π–π stacking interactions of 1 and 4 are 142.2 and 19.6 meV, respectively. From the orbital interactions depicted in Fig. 7, we can rationalize the electronic couplings between the HOMOs. The π-orbitals in 1 are head-to-head interactions, resulting in enhanced coupling by the overlap of orbitals. While the π-orbitals in 2 are mismatched, leading to the noneffective orbital overlap or partial cancellation between the wavefuctions of π-orbitals.
 | ||
| Fig. 6 Charge hopping pathways for compounds 1, 2, 4, 5, 5a and 5b. | ||
| Compd | Pathway | d (Å) | Vhab (meV) | Veab (meV) |
|---|---|---|---|---|
| a Data in parentheses obtained from ref. 24 and 58. | ||||
| 1 | 1, 2 | 3.88 | 142.2 | 44.4 |
| 3, 4, 5, 6 | 5.88 | 0.3 | 20.5 | |
| 2 | 1 | 3.74 | 57.8 (62.2)a | 23.1 |
| 2, 5 | 6.29 | 19.9 (2.7) | 76.1 | |
| 3 | 6.75 | 2.8 (2.0) | 12.4 | |
| 4, 6 | 5.16 | 3.9 (7.4) | 64.4 | |
| 4 | 1, 2 | 4.72 | 19.6 | 34.4 |
| 3, 4 | 6.20 | 0.1 | 2.3 | |
| 5, 6 | 7.79 | 6.4 | 17.4 | |
| 5 | 1, 2 | 5.74 | 15.5 (11.3) | 8.9 |
| 3, 4, 5, 6 | 4.77 | 33.0 (35.4) | 26.7 | |
| 5a | 1, 2 | 6.17 | 21.0 | 7.8 |
| 3, 4, 5, 6 | 4.80 | 43.5 | 41.6 | |
| 5b | 1, 2 | 6.03 | 22.6 | 3.2 |
| 3, 4 | 9.41 | 4.5 | 2.5 | |
| 5, 6 | 12.25 | 0.7 | 1.3 | |
| 7, 8 | 13.22 | 1.0 | 2.0 | |
 | ||
| Fig. 7 The dimer geometries and pictorial HOMO interactions of π–π stacking configurations in 1 and 4 viewed along the long molecular axis ((a) and (c)) and the short molecular axis ((b) and (d)), respectively. | ||
The crystal structures of the dimers of β-trithiophene 2 and 5 exhibit the respective sandwich-herringbone and herringbone arrangement. There is a π-stacking pathway in 2 which gives a maximum electronic coupling for hole of 57.8 meV, which is in consistent with previous calculated results (62.2 meV).24,58 For compound 5, the calculated electronic couplings for a hole for two kinds of pathways are 15.5 and 33.0 meV respectively. The introduction of phenyl or thienyl groups at the longitudinal ends of compound 5 lead to distinct molecular stacking in the crystal. Compound 5a has a herringbone arrangement, which is similar to that of 5. The existence of strong intermolecular phenyl–phenyl and phenyl–trithiophene interactions rationalize the large electronic couplings of 21.0 and 43.5 meV for a hole. The crystal structure of 5b contains multiple interactions, such as slipped π–π interactions and S⋯S interactions, however, the electronic couplings are relatively small due to the large intermolecular distances.
From the crystal structure, we notice that the short S⋯S contacts could make the molecules closely packed in the crystal structure. Furthermore such S⋯S interactions could effectively facilitate electron transport because of the high polarizability of sulfur atoms. Selected short S⋯S contacts in compounds 1, 2, and 5 and LUMO interactions are shown in Fig. 8. Compound 1 has multiple intensive S⋯S contacts, which are smaller than the sum of the van der Waals radius of S atoms (3.70 Å), leading to strong electronic coupling of 20.5 meV for an electron. In compound 2 and 5, strong S⋯S interactions (3.36 Å and 3.57 Å) are responsible for their large electronic couplings of 76.1 and 26.7 meV for an electron, respectively. Such S⋯S interactions could be in favor of electron transport.
 | ||
| Fig. 8 Pictorial LUMO interactions for short S⋯S contacts for 1, 2, and 5. | ||
It has been proved that the anisotropic transport properties would be prominent because of different molecular arrangements in different directions in the crystal. When the charge transport is dominant within a two-dimensional molecular layer and less efficient between molecular layers, the angular resolution anisotropic mobility within a molecular layer can be predicted by the following formula:66
 | (5) |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) cos2(θi − ϕ) is the relative hopping probability of various transport pathways to the specific transistor channel. As exemplified in Fig. 9a for 5, ϕ is the orientation angle of the conducting channel relative to the crystallographic axis c and θi are the angles of different pathways relative to the reference axis. θi − ϕ is the angle between the different pathways and the conducting channel. The anisotropic mobilities for a hole in 5 and 5a are shown in Fig. 9b. We can see clearly the hole mobilities vary with ϕ. The hole mobility has maximum values respectively of 0.066 cm2 V−1 s−1 and 0.045 cm2 V−1 s−1 for 5a and 5 with ϕ of 90°. The anisotropic mobilities for the available crystal structures of symmetric compounds in specific layers are collected in Table 5, which give the mobility ranges for different directions within a crystal plane.
cos2(θi − ϕ) is the relative hopping probability of various transport pathways to the specific transistor channel. As exemplified in Fig. 9a for 5, ϕ is the orientation angle of the conducting channel relative to the crystallographic axis c and θi are the angles of different pathways relative to the reference axis. θi − ϕ is the angle between the different pathways and the conducting channel. The anisotropic mobilities for a hole in 5 and 5a are shown in Fig. 9b. We can see clearly the hole mobilities vary with ϕ. The hole mobility has maximum values respectively of 0.066 cm2 V−1 s−1 and 0.045 cm2 V−1 s−1 for 5a and 5 with ϕ of 90°. The anisotropic mobilities for the available crystal structures of symmetric compounds in specific layers are collected in Table 5, which give the mobility ranges for different directions within a crystal plane.
 | ||
| Fig. 9 The angle-dependent hopping paths projected to a transistor channel in the bc plane (a) and calculated angle-resolved anisotropic hole mobilities (b) of 5 and 5a, respectively. | ||
| Theo. | Aniso. | Expt. | |||
|---|---|---|---|---|---|
| μh | μe | μh | μe | μ | |
| a Ref. 22 and 23.b Ref. 21 and 24.c Ref. 21.d Ref. 73.e Ref. 20. | |||||
| 1 | 0.23 | 0.03 | 0–0.69 | 0.013–0.073 | 0.05a |
| 2 | 0.02 | 0.08 | 0.0016–0.061 | 0.057–0.20 | 0.005b |
| 4 | 0.0087 | 0.052 | 0–0.026 | 0.013–0.12 | 0.08c |
| 5 | 0.023 | 0.018 | 0.028–0.045 | 0.020–0.035 | 0.89a |
| 5a | 0.039 | 0.074 | 0.051–0.066 | 0.091–0.13 | 2.0d |
| 5b | 0.022 | 0.0086 | 0–0.068 | 0–0.026 | 0.002e |
The predicted average mobilities are listed in Table 5, together with the available experimental data. The calculated hole mobility of 1 is 10 times of that of 2, which is well consistent with the experimental values. Compound 1 prefers transporting holes because of a relatively smaller reorganization energy and larger electronic couplings for a hole. While compound 2 prefers electron transport since the existence of multiple short S⋯S interaction enhances the electronic couplings for an electron. The predicted hole mobilities for 5 and 5a are slightly larger than that of 2 and much smaller than experimental values. The vinylene bridge in compounds 5 and 5a decreases the reorganization energy, the hole injection barrier and aromaticity, which could be beneficial to increase the hole mobility. Considering the fact that the experimental mobilities are strongly influenced by the microstructural characteristics of the dielectric layers in OFETs, such as film deposition temperature, film growth mode, and semiconductor phase composition,72 our calculated mobility based on the crystal structures can be regarded as a reference.
Conclusions
In summary, the electronic and charge transport properties of dimers of dithienothiophenes have been investigated by DFT calculations. The frontier orbitals of the dimers have the same symmetry as the molecular symmetry, indicating that the molecular symmetry determines the electron density distribution in the molecules. The symmetries of the dimers also influence the molecular arrangements in the crystal states. The vinylene-bridged dimers have relatively smaller reorganization energies, more matchable HOMO and LUMO levels to the electrode of the device, decreased aromaticities and larger mobilities in comparison to single-bond linked dimers. So both the symmetry and linking modes are important to determine the electronic and charge transport properties of dimers of dithienothiophenes.The calculated results would hint us that a highly symmetrical molecule could have an advantage in the molecular arrangement in the solid state. The extended π conjugation could enhance the electronic couplings through π–π stacking interactions, meanwhile, the decreased molecular aromaticity could be more favorable for charge transport. The highly symmetrical vinylene-bridged dimers of dithienothiophenes, or their analogues, could be potentially good candidates for transistor applications.
Acknowledgements
We are grateful for the financial support from the National Nature Science Foundation of China (grant no. 21173101 and 21073077).References
- J. E. Anthony, Chem. Rev., 2006, 106, 5028–5048 CrossRef CAS PubMed.
- V. Coropceanu, J. Cornil, D. A. da Silva Filho, Y. Oliver, R. Silbey and J. L. Bredas, Chem. Rev., 2007, 107, 926–952 CrossRef CAS PubMed.
- J. Zaumseil and H. Sirringhaus, Chem. Rev., 2007, 107, 1296–1323 CrossRef CAS PubMed.
- J. E. Anthony, Angew. Chem., Int. Ed., 2008, 47, 452–483 CrossRef CAS PubMed.
- Y. Wen, Y. Liu, Y. Guo, G. Yu and W. Hu, Chem. Rev., 2011, 111, 3358–3406 CrossRef CAS PubMed.
- M. Mas-Torrent and C. Rovira, Chem. Rev., 2011, 111, 4833–4856 CrossRef CAS PubMed.
- C. Wang, H. Dong, W. Hu, Y. Liu and D. Zhu, Chem. Rev., 2012, 112, 2208–2267 CrossRef CAS PubMed.
- T. M. Clarke and J. R. Durrant, Chem. Rev., 2010, 110, 6736–6767 CrossRef CAS PubMed.
- J. E. Allen and C. T. Black, ACS Nano, 2011, 5, 7986–7991 CrossRef CAS PubMed.
- T. Ameri, N. Li and C. J. Brabec, Energy Environ. Sci., 2013, 6, 2390–2413 CAS.
- S.-C. Lo and P. L. Burn, Chem. Rev., 2007, 107, 1097–1116 CrossRef CAS PubMed.
- S. Chen, L. Deng, J. Xie, L. Peng, L. Xie, Q. Fan and W. Huang, Adv. Mater., 2010, 22, 5227–5239 CrossRef CAS PubMed.
- H. Sasabe and J. Kido, J. Mater. Chem. C, 2013, 1, 1699–1707 RSC.
- T. Otsubo, Y. Aso and K. Takimiya, J. Mater. Chem., 2002, 12, 2565–2575 RSC.
- A. Mishra, C.-Q. Ma and P. Bäuerle, Chem. Rev., 2009, 109, 1141–1276 CrossRef CAS PubMed.
- X. Zhang, A. P. Côté and A. J. Matzger, J. Am. Chem. Soc., 2005, 127, 10502–10503 CrossRef CAS PubMed.
- R. M. Osuna, X. Zhang, A. J. Matzger, V. Hernndez and J. T. Lpez Navarrete, J. Phys. Chem. A, 2006, 110, 5058–5065 CrossRef CAS PubMed.
- X. Zhang, J. P. Johnson, J. W. Kampf and A. J. Matzger, Chem. Mater., 2006, 18, 3470–3476 CrossRef CAS.
- Y. M. Sun, Y. Q. Ma, Y. Q. Liu, Y. Y. Lin, Z. Y. Wang, Y. Wang, C. A. Di, K. Xiao, X. M. Chen, W. F. Qiu, B. Zhang, G. Yu, W. P. Hu and D. B. Zhu, Adv. Funct. Mater., 2006, 16, 426–432 CrossRef CAS PubMed.
- W. Jiang, Y. Li and Z. Wang, Chem. Soc. Rev., 2013, 42, 6113–6127 RSC.
- L. Zhang, L. Tan, W. Hu and Z. Wang, J. Mater. Chem., 2009, 19, 8216–8222 RSC.
- H. Sirringhaus, R. H. Friend, X. C. Li, S. C. Moratti, A. B. Holmes and N. Feeder, Appl. Phys. Lett., 1997, 71, 3871–3873 CrossRef CAS PubMed.
- X.-C. Li, H. Sirringhaus, F. Garnier, A. B. Holmes, S. C. Moratti, N. Feeder, W. Clegg, S. J. Teat and R. H. Friend, J. Am. Chem. Soc., 1998, 120, 2206–2207 CrossRef CAS.
- L. Tan, L. Zhang, X. Jiang, X. Yang, L. Wang, Z. Wang, L. Li, W. Hu, Z. Shuai, L. Li and D. Zhu, Adv. Funct. Mater., 2009, 19, 272–276 CrossRef CAS PubMed.
- M. C. R. Delgado, K. R. Pigg, D. A. da Silva Filho, N. E. Gruhn, Y. Sakamoto, T. Suzuki, R. M. Osuna, J. Casado, V. Hernández, J. T. L. Navarrete, N. G. Martinelli, J. Cornil, R. S. Sánchez-Carrera, V. Coropceanu and J.-L. Brédas, J. Am. Chem. Soc., 2009, 131, 1502–1512 CrossRef CAS PubMed.
- C. L. Wang, F. H. Wang, Q. K. Li and Z. G. Shuai, Org. Electron., 2008, 9, 635–640 CrossRef CAS PubMed.
- S.-H. Wen, W.-Q. Deng and K.-L. Han, Phys. Chem. Chem. Phys., 2010, 12, 9267–9275 RSC.
- A. Troisi, Chem. Soc. Rev., 2011, 40, 2347–2358 RSC.
- H. Li, R. Zheng and Q. Shi, Phys. Chem. Chem. Phys., 2011, 13, 5642–5650 RSC.
- Y. Geng, J. Wang, S. Wu, H. Li, F. Yu, G. Yang, H. Gao and Z. Su, J. Mater. Chem., 2011, 21, 134–143 RSC.
- H. Liu, S. Kang and J. Y. Lee, J. Phys. Chem. B, 2011, 115, 5113–5120 CrossRef CAS PubMed.
- L. Wang, P. Li, B. Xu, H. Zhang and W. Tian, Org. Electron., 2014, 15, 2476–2485 CrossRef CAS PubMed.
- X. D. Yang, L. J. Wang, C. L. Wang, W. Long and Z. G. Shuai, Chem. Mater., 2008, 20, 3205–3211 CrossRef CAS.
- X. D. Yang, Q. K. Li and Z. G. Shuai, Nanotechnology, 2007, 18, 424029 CrossRef PubMed.
- Y. C. Cheng, R. J. Silbey, D. A. dasilva Filho, J. P. Calbert, J. Cornil and J. L. Brédas, J. Chem. Phys., 2003, 118, 3764–3774 CrossRef CAS PubMed.
- A. Troisi and G. Orlandi, J. Phys. Chem. B, 2005, 109, 1849–1856 CrossRef CAS PubMed.
- K. Sakanoue, M. Motoda, M. Sugimoto and S. Sakaki, J. Phys. Chem. A, 1999, 103, 5551–5556 CrossRef CAS.
- J.-L. Brédas, D. Beljonne, V. Coropceanu and J. Cornil, Chem. Rev., 2004, 104, 4971–5004 CrossRef PubMed.
- W. Q. Deng and W. A. Goddard III, J. Phys. Chem. B, 2004, 108, 8614–8621 CrossRef CAS.
- C. T. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- N. M. O’Boyle, A. L. Tenderholt and K. M. Langner, J. Comput. Chem., 2008, 29, 839–845 CrossRef PubMed.
- R. A. Marcus, Rev. Mod. Phys., 1993, 65, 599 CrossRef CAS.
- R. A. Marcus, Annu. Rev. Phys. Chem., 1964, 15, 155–196 CrossRef CAS.
- Y. A. Berlin, G. R. Hutchison, P. Rempala, M. A. Ratner and J. Michl, J. Phys. Chem. A, 2003, 107, 3970–3980 CrossRef CAS.
- B. S. Brunschwig, J. Logan, M. D. Newton and N. Sutin, J. Am. Chem. Soc., 1980, 102, 5798–5809 CrossRef CAS.
- J. E. Norton and J.-L. Brdas, J. Am. Chem. Soc., 2008, 130, 12377–12384 CrossRef CAS PubMed.
- I. Vilfan, Phys. Status Solidi B, 1973, 59, 351–360 CrossRef CAS PubMed.
- G. R. Hutchison, M. A. Ratner and T. J. Marks, J. Am. Chem. Soc., 2005, 127, 2339–2350 CrossRef CAS PubMed.
- H. Gao, C. Qin, H. Zhang, S. Wu, Z.-M. Su and Y. Wang, J. Phys. Chem. A, 2008, 112, 9097–9103 CrossRef CAS PubMed.
- T. Fujita, H. Nakai and H. Nakatsuji, J. Chem. Phys., 1996, 104, 2410–2417 CrossRef CAS PubMed.
- A. Troisi and G. Orlandi, J. Phys. Chem. A, 2006, 110, 4065–4070 CrossRef CAS PubMed.
- S. W. Yin, Y. P. Yi, Q. X. Li, G. Yu, Y. G. Liu and Z. G. Shuai, J. Phys. Chem. A, 2006, 110, 7138–7143 CrossRef CAS PubMed.
- B. M. Wong, M. Piacenza and F. D. Sala, Phys. Chem. Chem. Phys., 2009, 11, 4498–4508 RSC.
- N. Martsinovich and A. Troisi, Energy Environ. Sci., 2011, 4, 4473–4495 CAS.
- Y. Yi, L. Zhu and J.-L. Brdas, J. Phys. Chem. C, 2012, 116, 5215–5224 CAS.
- L. J. Wang, G. J. Nan, X. D. Yang, Q. Peng, Q. K. Li and Z. G. Shuai, Chem. Soc. Rev., 2010, 39, 423–434 RSC.
- J. Shi, L. Xu, Y. Li, M. Jia, Y. Kan and H. Wang, Org. Electron., 2013, 14, 934–941 CrossRef CAS PubMed.
- V. T. T. Huong, H. T. Nguyen, T. B. Tai and M. T. Nguyen, J. Phys. Chem. C, 2013, 117, 10175–10184 CAS.
- P. v. R. Schleyer, M. Manoharan, Z.-X. Wang, B. Kiran, H. Jiao, R. Puchta and N. J. R. V. E. Hommes, Org. Lett., 2001, 3, 2465–2468 CrossRef CAS PubMed.
- T. M. Krygowski, J. Chem. Inf. Comput. Sci., 1993, 33, 70–78 CrossRef CAS.
- M.-Y. Kuo, H.-Y. Chen and I. Chao, Chem.–Eur. J., 2007, 13, 4750–4758 CrossRef CAS PubMed.
- L. Wang and H. Zhang, J. Phys. Chem. C, 2011, 115, 20674–20681 CAS.
- L. Wang, G. Duan, Y. Ji and H. Zhang, J. Phys. Chem. C, 2012, 116, 22679–22686 CAS.
- J.-D. Huang, S.-H. Wen, W.-Q. Deng and K.-L. Han, J. Phys. Chem. B, 2011, 115, 2140–2147 CrossRef CAS PubMed.
- V. I. Minkin, M. N. Glukhovtsev and B. Y. Simkin, Aromaticity and Antiaromaticity, Wiley, New York, 1994 Search PubMed.
- T. M. Krygowski, H. Szatylowicz, O. A. Stasyuk, J. Dominikowska and M. Palusiak, Chem. Rev., 2014, 114, 6383–6422 CrossRef CAS PubMed.
- W. B. Chen, H. X. Li, J. R. Widawsky, C. Appayee, L. Venkataraman and R. Breslow, J. Am. Chem. Soc., 2014, 136, 918–920 CrossRef CAS PubMed.
- M. L. Tang, J. H. Oh, A. D. Reichardt and Z. Bao, J. Am. Chem. Soc., 2009, 131, 3733–3740 CrossRef CAS PubMed.
- J. L. Brdas, J. P. Calbert, D. A. da Silva Filho and J. Cornil, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5804–5809 CrossRef PubMed.
- C. Kim, A. Facchetti and T. Marks, Adv. Mater., 2007, 19, 2561–2566 CrossRef CAS PubMed.
- L. Zhang, L. Tan, Z. H. Wang, W. P. Hu and D. B. Zhu, Chem. Mater., 2009, 21, 1993–1999 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2015 |