
Catalytically engineered reduced graphene oxide/ZnO hybrid nanocomposites for the adsorption, photoactivity and selective oil pick-up from aqueous media†
K. B. Babitha,
J. Jani Matilda,
A. Peer Mohamed and
S. Ananthakumar*
Functional Materials Section, Materials Science and Technology Division, CSIR-National Institute for Interdisciplinary Science and Technology, Thiruvananthapuram, Kerala, India. E-mail: ananthakumar70@gmail.com; ananthakumars@niist.res.in; Fax: +91 471 2491712; Tel: +91 471 2515289
First published on 21st May 2015
Abstract
Exfoliated graphene oxide (GO) is catalytically activated via in situ growth of nanocrystalline ZnO crosslinked with 3-aminopropyltrimethoxy silane (APTMS) to result in ZnO/Si@rGO hybrid nanocomposite architectures. Structural and morphological features are systematically ascertained using XRD, SEM, TEM, TGA, UV-vis, FTIR and BET techniques. ZnO/Si@rGO hybrid nanocomposites exhibit a high surface area (78 m2 g−1). It is significantly large compared to phase pure nano-ZnO where the surface area is only 18 m2 g−1. Hybrid nanocomposite also shows ∼55% photocatalytic activity and ∼59% adsorption of dye from aqueous system containing 340 μM methylene blue (MB) dye. It was compared with pure ZnO, which shows only ∼0% adsorption and ∼31% photocatalytic activity. In the similar way, ZnO@rGO nanocomposites in the absence of a crosslinking agent were prepared and characterized. The adsorption and photocatalytic degradation property of both ZnO@rGO and ZnO/Si@rGO against the adsorption and photodegradation of a series of cationic organic dyes were compared for the first time. Interestingly, when ZnO/Si@rGO hybrid nanocomposite is deposited onto the surface of a cotton textile, it showed an excellent hydrophobic surface with a contact angle of 113.7°. Cotton textile modified with hybrid nanocomposite was explored further for the selective pick-up of engine oil contaminants from the aqueous media. The combined properties of dye adsorption, photodegradation and oil sorption properties of ZnO/Si@rGO hybrid nanocomposite enable the design of novel self-regenerative catalytically active cotton textile filters. A facile UV bleaching retrieves the cotton substrate for continuous use. In addition to this, ZnO–rGO composites were also prepared by a physical blending method and compared with ZnO/Si@rGO hybrid nanocomposite and ZnO@rGO nanocomposite.
1. Introduction
Design of functionally activated catalytic hybrid materials has received great attention in recent years with special focus on environmental remediation applications.1,2 Graphene oxide (GO), a derivative of graphite oxide, is composed of either a single or a few layers of graphene sheets. It offers many oxygenated surface hydrophilic functionalities preferred much for the sorption as well as membrane applications due to its meso/nano porous layered microstructures, ready dispersion in aqueous/organic media, resistance against bacterial activity and tunable surface functionality.3,4 2D GO nanosheets have been demonstrated for the adsorption of Pb(II), Co(II) and Cd(II) heavy metal ions.5 Wang et al.6 and Zhang et al.7 conducted the adsorption of polycyclic aromatic hydrocarbons such as naphthalene, phenanthrene and pyrene on graphene nanostructures. Yang et al. proposed GO–Fe2O3 nanocomposites for the adsorption of organic and inorganic pollutants.8 These reports strongly indicate the nanostructured GO is a potential sorbent for many hazardous chemical pollutants. Researchers made attempts to catalytically modify GO/reduced graphene oxide (rGO) in order to chemically decompose the hazardous pollutants and subsequently regenerate the material for any continuous use. It is achieved via in situ growth of catalytic grade semiconducting metal oxide nanostructures that successfully introduced dual-functionality; simultaneous adsorption and degradation of organic/inorganic contaminants and self-regeneration capacity.In this context, the design of GO nanocomposite architectures involving catalytically active nano crystalline TiO2, CdS, and ZnO has received prime importance. Gu et al. have grown anatase nano-TiO2 on rGO nanosheets and reported that rGO/TiO2 hybrid had better photoactivity compared to its nano counterpart.9 Jia et al. reported (N-graphene)/CdS nanocomposite as co-catalyst for photocatalytic H2 production.10 Zhang et al. developed CdS–graphene–TiO2 hybrids through in situ strategy on the flatland of GO for the selective photocatalytic oxidation of alcohols to aldehydes.11 An enhanced visible light photocatalytic performance of graphene/TiO2 nanocomposites is also reported in this series.12 It is clear from these reports that rGO is extensively studied with photoactive TiO2 compared to its competitive ZnO/rGO architectures. ZnO has poor surface area, and it is prone to electron–hole recombination, which limits its use as an efficient photocatalyst. The growth of rGO supported ZnO nanocatalyst can partially resolve these limitations.13
In this work, in situ synthesis of APTMS crosslinked ZnO/Si@rGO hybrid nanocomposite is designed via microwave reflux technique, and the beneficial properties of hybrid architectures are studied for the adsorption, photodegradation and surface hydrophobic properties. The performance of hybrid nanocatalyst is compared with the ZnO@rGO nanocomposite and physically blended ZnO–rGO. A cotton textile filter was designed using the ZnO/Si@rGO hybrid nanocomposites and tested for the selective adsorption of oil contaminants from aqueous medium. Comparative studies with respect to the physiochemical properties, adsorption as well as photocatalytic degradation of catalytically modified ZnO@rGO, ZnO/Si@rGO and ZnO–rGO architectures were systematically attempted and reported for the first time.
2. Experimental section
2.1 Materials
Graphite was procured from Sigma Aldrich, Germany (99.9%). Sodium nitrate (NaNO3, 99.5%), potassium permanganate (KMnO4, 99%) and hydrogen peroxide (H2O2, 99%) were obtained from S. D. Fine Chemicals Limited, India. Zinc nitrate hexahydrate (Zn(NO3)3·6H2O, 99%) and conc. sulphuric acid (H2SO4, 99%) were supplied by Merck, India. Lithium hydroxide (LiOH, 99%), methylene blue (MB) (C16H18ClN3S) and crystal violet (CV) (C25H30ClN3) were purchased from SRL, India. Malachite green (MG) (C23H25ClN2) and rhodamine 6G (R6G) (C28H31ClN2O3) were obtained from Qualigen Fine Chemicals, India, and S. D. Fine Chemicals Limited, India, respectively. All the reagents were used as received without any further purification.2.2 Synthesis of GO
GO was synthesized by the oxidation and exfoliation of powder graphite (GR) via a well reported modified Hummers method.14 In a typical experiment, 1 g GR, 1 g NaNO3 and 50 mL conc. H2SO4 were successively added into a beaker and stirred magnetically for 30 min in an ice bath. To this mixture, 3 g of KMnO4 was added and the resultant mixture was transferred to a water bath maintained at 35 °C. The whole reaction mixture was diluted with 150 mL of distilled water. In this diluted mass, 10 mL of 30% H2O2 was gradually introduced. After vigorous stirring for 60 min, the end product was centrifuged and purified by repeated washing with double distilled water until the pH became 7. Finally, exfoliated GO was obtained by vacuum drying at 65 °C.2.3 Preparation of ZnO/rGO nanoarchitectures (ZnO@rGO, ZnO/Si@rGO and ZnO–rGO)
A desired amount of GO was dispersed in aqueous medium and sonicated for 30 min. To this mixture, 0.2 M Zn(NO3)3·6H2O solution was added under ultrasonication by keeping the GO/ZnO weight (wt) ratio 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. The reaction mixture was subsequently treated with 10 wt% LiOH for the in situ precipitation of Zn(OH)2 at room temperature. The pH of the reaction was maintained as ∼8.5. A homogeneous dispersion of GO deposited with Zn(OH)2 was obtained, which was taken to the microwave synthesis work station (Sineo, MAS II) operating at a frequency of 2.45 GHz and having a microwave power in the range 0–1000 W. A constant temperature of 80 °C was maintained for the microwave refluxing and the reactant mixture was refluxed for 30 min at microwave power 300 W. Microwave irradiated ZnO modified rGO was finally centrifuged and washed with double distilled water until the pH of the filtrate became neutral. The ZnO@rGO nanocomposite product was then vacuum-dried at 65 °C.
:1. The reaction mixture was subsequently treated with 10 wt% LiOH for the in situ precipitation of Zn(OH)2 at room temperature. The pH of the reaction was maintained as ∼8.5. A homogeneous dispersion of GO deposited with Zn(OH)2 was obtained, which was taken to the microwave synthesis work station (Sineo, MAS II) operating at a frequency of 2.45 GHz and having a microwave power in the range 0–1000 W. A constant temperature of 80 °C was maintained for the microwave refluxing and the reactant mixture was refluxed for 30 min at microwave power 300 W. Microwave irradiated ZnO modified rGO was finally centrifuged and washed with double distilled water until the pH of the filtrate became neutral. The ZnO@rGO nanocomposite product was then vacuum-dried at 65 °C.
Similarly, identical procedure was followed for the synthesis of ZnO/Si@rGO hybrid nanocomposites by taking 1:1 ZnO/GO (wt ratio) composition. The hybrid nanocomposite was formed by the in situ addition of APTMS. Silane/ZnO molar ratio of 0.3 was maintained. For comparison, a ZnO–rGO composite mixture was further prepared by physical blending. Nano-ZnO was synthesized via a microwave assisted precipitation of 0.2 M Zn(NO3)2·6H2O using LiOH. Without any heat treatment the nano-ZnO was physically blended with an equal amount of rGO to obtain ZnO–rGO with wt ratio 1:1. A schematic of ZnO@rGO nanocomposite, ZnO/Si@rGO hybrid nanocomposite and ZnO–rGO composite preparation is shown in Scheme 1.
 | ||
| Scheme 1 Illustration of the processing of different ZnO/rGO nanoarchitectures (A) ZnO@rGO, (B) ZnO/Si@rGO and (C) ZnO–rGO. | ||
2.4 Fabrication of ZnO/Si@rGO sorbent coated cotton textile
A cotton textile (2 cm × 1 cm) was immersed in double distilled water under gentle magnetic stirring. The textile was squeezed to remove excess water and dried in the oven at 80 °C. For coating, ZnO/Si@rGO hybrid nanocomposite dispersion (0.4 wt%) was prepared in aqueous medium, and the cotton textile was soaked for three times for the firm deposition of ZnO/Si@rGO hybrid nanocomposite. Finally, the cotton substrate was washed in double distilled water to remove loosely fixed particles. It was then allowed to dry overnight in the oven under ambient conditions.152.5 Characterizations
Structural composition and phase purity was analyzed using X-ray diffractogram (XRD, X' Pert Pro Philips) equipped with a Cu Kα source, operating in the diffraction angle (2θ) of 10–60°. The average crystallite size of the particles was calculated using Debye–Scherrer equation as DXRD = kλ/βcosθ, where DXRD is the average crystallite size in nm, k is the shape factor (0.9), λ is the wavelength in nm, β is the full width at half maximum of (101) peak in radians and θ is the diffraction angle (degree).16 Microstructures and morphological features were monitored using a scanning electron microscope (SEM, ZEISS EVO 18) operated at 20 kV. The chemical constituents of the samples were obtained from the elemental diffraction X-ray (EDX) analysis mounted with the SEM instrument. The transmission electron microscopic (TEM) images and high resolution TEM (HRTEM) images along with selected area electron diffraction (SAED) patterns were obtained using Tecnai G2, FEI, Netherlands, transmission electron microscope operating at an accelerating voltage 300 kV. For TEM analysis, samples were prepared by evaporating a drop of diluted sample dispersion prepared in acetone on a copper grid. Thermal stability of ZnO@rGO nanocomposite, ZnO/Si@rGO hybrid nanocomposite and ZnO–rGO composite were seen using Shimadzu TG-50 thermogravimetric analyzer (TGA) from room temperature to 1000 °C at a heating rate of 10 °C min−1 in a constant flow of O2. Optical properties were analyzed using a UV-vis spectrophotometer (UV-2401 PC, Shimadzu) and the spectra were recorded in the range from 200 to 600 nm. The direct band was calculated by equation (αhν)2 = A(hν − Eg), where α is the absorption coefficient, hν is the photon energy, Eg is the optical band gap in eV and A is a constant. A plot of (αhν)2 against hν gives the band gap energy, which was obtained by extrapolating the tangent of the curve to (αhν)2 = 0.17 Fourier transform infrared analysis (FTIR) was carried out using an IR Prestige 21 (SHIMADZU) spectrometer at a scanning range of 400 to 4000 cm−1. Surface area and pore size distribution analysis was carried out using Brunauer–Emmet–Teller (BET) method using the instrument (Micromeritics Gemini 2375 Surface Area Analyzer) after degassing the powder for 2 h at 150 °C. Contact angle measurements of the bare cotton textile and ZnO/Si@rGO coated cotton textile were conducted with dynamic contact angle meter and tensiometer data physics.
2.6 Dye adsorption and photocatalytic activity studies
The adsorption and degradation experiments were conducted with UV-vis spectroscopy by measuring the absorbance from 200 to 800 nm. The concentration of the dye in the solution was determined by Beer–Lambert law expression. Because the concentration of the dye used was high (340 μM), the dye samples were diluted before absorbance measurements. Batch adsorption as well as degradation experiments were conducted. Preliminary experiments indicated that the adsorption as well as degradation of dye reached equilibrium in ∼1 h. Thus, the contact time of 2 h was given for all the batch experiments. All experiments were conducted in duplicate, and only the mean values were reported. Blank experiments without the addition of catalyst were also conducted to ensure that the decrease in the concentration was actually due to the adsorption as well as degradation of catalytically activated rGO architectures rather than by the adsorption on the glass bottle wall.The adsorption experiments were performed by adding a specified amount of the ZnO/rGO nanoarchitectures (0.4 g L−1) to the beakers containing 75 mL of 340 μM dye solutions each. The aqueous suspension was stirred continuously under dark conditions. A small amount of aliquots was collected at different time intervals and centrifuged to separate the dye adsorbed powder. The remaining concentration of the dye was measured from the absorbance peak before and after the adsorption by UV-vis absorption spectroscopy. The characteristic absorption band of MB, MG, CV and R6G occurred at 656, 620, 590 and 530 nm, respectively. The % of dye adsorption was calculated using the equation, % of adsorption = [(C0 − C)/C0]100, where C0 (mg L−1) and C (mg L−1) are the dye concentrations at the initial time and after adsorption at time t, respectively.18
The photocatalytic activity of the ZnO/rGO nanoarchitectures was evaluated from the photodegradation of 340 μM dye solution in UV radiation. For a typical experiment, 0.4 g L−1 catalyst powder was added to 75 mL of 340 μM dye solution and stirred continuously for 30 min under dark conditions in order to obtain stabilisation of dye solution over the catalyst powder subsequently exposed to UV radiation. Aliquots were collected at different time intervals, and the same procedure was adopted.19 The % of dye degradation was calculated using the equation, dye degradation (%) = [C/C0]100. The photocatalytic degradation follows the Langmuir–Hinshelwood mechanism,20 which may be represented as dC/dt = kappC, where, dC/dt is the rate of change of MB concentration with respect to UV irradiation time t. kapp is the apparent first order reaction rate constant (min−1), and C is the concentration of MB dye at time t. The solution obtained when integrating the above equation is ln[C0]/[C] = kappt, where C0 is the initial concentration of the MB dye solution. The plot of ln[C0]/[C] against UV irradiation time t gives a straight line. kapp was obtained from the slope of ln[C0]/[C] against t. In the present work, adsorption and photodegradation of MB, MG, CV and R6G dyes were analysed using ZnO/rGO nanoarchitectures.
2.7 Oil adsorption study
The oil sorption property of the cotton textile modified with ZnO/Si@rGO hybrid nanocomposite in terms of engine oil was determined as follows. 0.4 g of cotton textile coated with ZnO/Si@rGO hybrid nanocomposite was placed in an oil-water system for 10 min. After removing excess oil, the weight of oil adsorbed ZnO/Si@rGO modified cotton textile was taken. The oil sorption capacity (g g−1) was calculated using the equation (S − S0)/S0, where S is the weight of the ZnO/Si@rGO modified cotton textile after oil uptake (g) and S0 is the initial weight of ZnO/Si@rGO modified cotton textile before oil uptake (g).21 In order to understand the efficiency of ZnO/Si@rGO, the same experiment was repeated with bare cotton textile. Experiments were conducted in duplicate and the mean values are reported.3. Results and discussion
3.1 Conversion of GR to GO
In Hummers method, KMnO4 and H2SO4 are used as oxidizing agents, where dimanganese heptoxide acts as the active species in the oxidation process.4 Adequate ultrasonication in water leads to the exfoliation of the oxidized layers. These exfoliated sheets usually contain one or a few layers of carbon atoms similar to graphene and resulted in GO. The conversion of GR to GO was established by various characterization tools. The powder X-ray diffraction analysis (Fig. 1A) shows the characteristic diffraction peak of hexagonal GR with a reflection peak centered at 26° (002). It also shows the crystalline nature of GO where the diffraction peak at 10.8° represents the (001) plane of GO.22,23 This indicates that GR is completely oxidized to GO sheets. The thermal stability of as-received GR measured by TG analysis is given in Fig. 1B. As seen in the figure, GR is stable up to 620 °C. A rapid and one step reduction in the weight is observed after 620 °C due to the decomposition of the graphitic carbon. The TG analysis of GO showed three step decompositions. The weight loss (∼5%) found below ∼100 °C is associated with the decomposition of physically adsorbed water molecules on the surface of hydrophilic GO sheets. The second weight loss of ∼8% between 200 and 250 °C is actually due to the loss of oxygen containing functional groups (e.g. –COOH and –O–) decorating the surface of GO. This was followed by a complete oxidative decomposition of the graphitic substrate in between 450 and 690 °C. The UV-vis absorption spectra of GO given in Fig. 1C describes the absorption maxima at 230 nm. It is consistent with the λmax value of GO reported in previous studies.24 A homogeneous dispersion of hydrophilic GO in water is also confirmed, and the photograph in the inset of Fig. 1C shows the stable GO dispersion. A high stability confirms the polarity of the GO surface.4 | ||
| Fig. 1 (A and B) X-ray diffraction analysis and TG analysis of GR and GO, respectively; (C) UV-vis absorption spectra of GO; (D–F) SEM image, TEM image and the SAED pattern of GR, respectively; (G–I) SEM image, TEM image and the SAED pattern of GO, respectively. The photograph showing the dispersion of GO in water is given in the inset of (C). | ||
The SEM image of GR, shown in Fig. 1D, describes sheet like GR with dimensions in the range 8–20 μm. The multilayered morphology of GR is confirmed from the TEM image shown in Fig. 1E. SEM image given in Fig. 1G shows micrometer sized GR sheets, which completely delaminated into submicron sized thin layers of GO with single or few layers. The TEM image of GO, given in Fig. 1H, displays exfoliated sheets of GO derived from GR. It appeared curly in nature.25 The crystalline nature of GR and GO were confirmed from the SAED patterns, which are given in Fig. 1F and I.
3.2 Characterization of ZnO/rGO nanoarchitectures
In the present work, we adopted microwave reflux technique for the synthesis of the ZnO/rGO nanoarchitectures. In the presence of a base, microwave heating resulted in the partial reduction of GO.26 The addition of APTMS resulted in the in situ anchoring of grown ZnO on the rGO sheets and resulted in the formation of ZnO/Si@rGO hybrid nanocomposites. The representative TEM images show the growth of well-defined nanocrystalline ZnO (Fig. S1A & B†). The TEM morphologies clearly show the advantage of rGO supported in situ growth because nucleation and growth of nanocrystalline ZnO is preferably occurring on the carbon layers of rGO sheets. In fact, rGO supported in situ growth resulted in size controlled nano-ZnO over rGO substrates. For example, in the absence of APTMS, the TEM and HRTEM images corresponding to ZnO@rGO (Fig. S1A† and 2A) showed that the rGO sheets are decorated with ∼10–20 nm sized ZnO nanoparticles. On the other hand, in the silane cross linked ZnO/Si@rGO hybrid nanocomposites, the rGO support contains a thin layer of evenly distributed ZnO with particle size below 3 nm (Fig. 2C). It indicates the capping effect of APTMS in controlling the ZnO growth.27 In case of ZnO–rGO composites prepared via a physical blending technique, the distribution of physically adhered, large sized ZnO with particle size ∼100 nm is seen over rGO sheets (Fig. S2A†). The characteristic particle size seen in TEM/HRTEM images proves that the rGO substrates are extremely helpful in suppressing the aggregation of ZnO nanocrystals. The SAED pattern shown in Fig. 2B explains the polycrystalline nature of ZnO modified rGO nanomaterials. In hybrid nanocomposites, the APTMS is found to reduce the crystalline nature slightly, which is expected (Fig. 2D). The EDX results as obtained from the SEM analysis are given in Fig. S1C and D.† In sample ZnO@rGO, the presence of C, O and Zn is evidenced. In ZnO/Si@rGO hybrid nanocomposite (Fig. S1D†), the additional peak of Si is noticed, which proves silane linkage between rGO and ZnO. | ||
| Fig. 2 (A and B) Representative HRTEM and SAED pattern ZnO@rGO; (C and D) HRTEM and SAED pattern of ZnO/Si@rGO. | ||
The XRD analysis, shown in Fig. 3A, gives the crystalline nature of ZnO@rGO, ZnO/Si@rGO and ZnO–rGO. The XRD patterns consist of all the characteristic peaks of ZnO at 2θ values of 32°, 34° and 36°, which are correspond to the (100), (101) and (002) planes. The diffraction peaks seen in ZnO@rGO, ZnO–rGO and ZnO/Si@rGO match well with the crystal structure of wurtzite ZnO. In addition to that, all the composites and hybrids show an additional peak at 26°, which clearly shows the conversion of GO to rGO under microwave irradiation. The ZnO crystallite sizes determined from the Debye Scherrer equation are 17, 17, 13 and 6 nm that can be put in the order of ZnO > ZnO–rGO > ZnO@rGO > ZnO/Si@rGO.
 | ||
| Fig. 3 (A) XRD patterns of ZnO, ZnO@rGO, ZnO–rGO and ZnO/Si@rGO. (B) TG analysis of ZnO, ZnO@rGO, ZnO–rGO and ZnO/Si@rGO. | ||
Fig. 3B shows the weight loss characteristics of ZnO, ZnO–rGO, ZnO@rGO and ZnO/Si@rGO grown in microwave irradiation. Among these, the APTMS capped hybrid nanocomposites have comparatively higher weight loss in the entire thermal heating range, which is actually contributed by the decomposition of organosilane and surface adsorbed nitrates. In this case, for ZnO/Si@rGO, a weight loss of ∼45% was observed in the temperatures 200–700 °C. ZnO–rGO and ZnO@rGO show similar weight loss up to 260 °C. At higher temperatures ZnO@rGO shows more weight loss than ZnO–rGO because of the presence of nitrates during the in situ formation of ZnO on rGO.
Results on the optical absorption properties of ZnO, ZnO–rGO, ZnO/Si@rGO and ZnO@rGO studied using UV-vis spectral analysis are presented in Fig. S3.† Freely grown bulk nano-ZnO has an on-set absorption band at 368 nm. It is red shifted to 375 nm when ZnO nanocrystals are grown on rGO supports. It is evident that there is a chemical interaction between rGO and catalytic nano-ZnO. The narrowing in band gap energy (3.15 eV to 3.00 eV) can demonstrate the significant influence of rGO on the optical characteristics.
Fig. 4A shows the FTIR analyses of ZnO, GO, ZnO@rGO and ZnO/Si@rGO. In the FTIR spectrum of ZnO, the absorption band at 527 cm−1 is attributed to the vibration of Zn–O bonds. The interaction of rGO shifted this peak to 490 cm−1 for ZnO@rGO and ZnO/Si@rGO. The stretching vibration of surface hydroxyl groups (–OH) is confirmed by the broad absorption band observed in between 3000 and 3400 cm−1.28 The bands corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O carbonyl stretching at 1726 cm−1 and CC stretching at 1617 cm−1 indicate the presence of GO in the sample.29 The IR spectrum of the ZnO/Si@rGO shows the presence of new bands at 1660 cm−1 and 1128 cm−1 corresponding to amide CO stretching and C–N bond stretching30 vibrations. This confirms the presence of the amide functional group formed due to the interaction of rGO and APTMS. For ZnO/Si@rGO, the absorption bands observed at 3310–3350 cm−1 are attributed to the stretching vibration of N–H bonds,31 which could be overlapped at 2930 cm−1, 2850 cm−1 and 1395 cm−1 corresponding to the asymmetric stretching, symmetric stretching32 and bending with the strong bands of –OH groups, respectively. The absorption band at vibrations33 of –CH2 groups are present in APTMS. The newly formed peak at 902 cm−1 shows the formation of the Zn–O–Si bond in the hybrid nanocomposite.34,35
O carbonyl stretching at 1726 cm−1 and CC stretching at 1617 cm−1 indicate the presence of GO in the sample.29 The IR spectrum of the ZnO/Si@rGO shows the presence of new bands at 1660 cm−1 and 1128 cm−1 corresponding to amide CO stretching and C–N bond stretching30 vibrations. This confirms the presence of the amide functional group formed due to the interaction of rGO and APTMS. For ZnO/Si@rGO, the absorption bands observed at 3310–3350 cm−1 are attributed to the stretching vibration of N–H bonds,31 which could be overlapped at 2930 cm−1, 2850 cm−1 and 1395 cm−1 corresponding to the asymmetric stretching, symmetric stretching32 and bending with the strong bands of –OH groups, respectively. The absorption band at vibrations33 of –CH2 groups are present in APTMS. The newly formed peak at 902 cm−1 shows the formation of the Zn–O–Si bond in the hybrid nanocomposite.34,35
 | ||
| Fig. 4 (A) FTIR spectra of ZnO, GO, ZnO@rGO and ZnO/Si@rGO. (B) Proposed schematic illustration showing the mechanism of ZnO/Si@rGO formation. | ||
Based on the FTIR analysis, the possible mechanisms for the synthesis of ZnO/Si@rGO hybrid nanocomposite are proposed and illustrated in Fig. 4B. APTMS is a known and well reported crosslinking agent. In the presence of water, APTMS undergoes hydrolysis and forms hydrolyzed APTMS (silanol). On partial condensation, these silanol oligomers co-condense with the surface hydroxyl groups of ZnO via Zn–O–Si bonds.34 Simultaneously, the –NH2 groups present in the APTMS interact with the epoxy group in rGO, and hence NH(CH2)3Si(OCH3)3 was attached to rGO.4 The absorption band of ZnO/Si@rGO obtained at 3310–3350 cm−1 agrees with this mechanism. Another possibility is that the interaction between –NH2 groups of APTMS and the –COOH groups of rGO sheets results in the formation of amide linkage. Consequently, ZnO grafted continuously on rGO sheets via Zn–O–Si bonds.
The N2 adsorption–desorption isotherms and the BET surface area values of the samples prepared in this work are given in Fig. 5A and B. Microwave strategy produced nano-ZnO with BET surface area of 18 m2 g−1. The modified Hummers route resulted in GO nanostructures having bulk surface area approximately 76.5 m2 g−1. In physically blended ZnO–rGO composite mixture, the surface area is decreased because of the preferential blocking of the pores by the large size nanocrystalline ZnO. It can be understood from the reduction in the mesopore volume of GO wherein the actual pore volume is decreased from 0.0957 cm2 g−1 to 0.000933 cm2 g−1. When the ZnO growth is developed in situ on the rGO, due to controlled size of ZnO nanocrystals, the ZnO@rGO nanocomposite gives improved surface area. In this case the surface area of 27.5 m2 g−1 is obtained. In hybrid nanocomposite appreciably high surface area is seen as an effect of APTMS capping. ZnO/Si@rGO hybrid nanocomposites have bulk surface area of 78 m2 g−1. It also has the mesopore volume as 0.101305 cm2 g−1, which is close to that of the GO nanosheets.
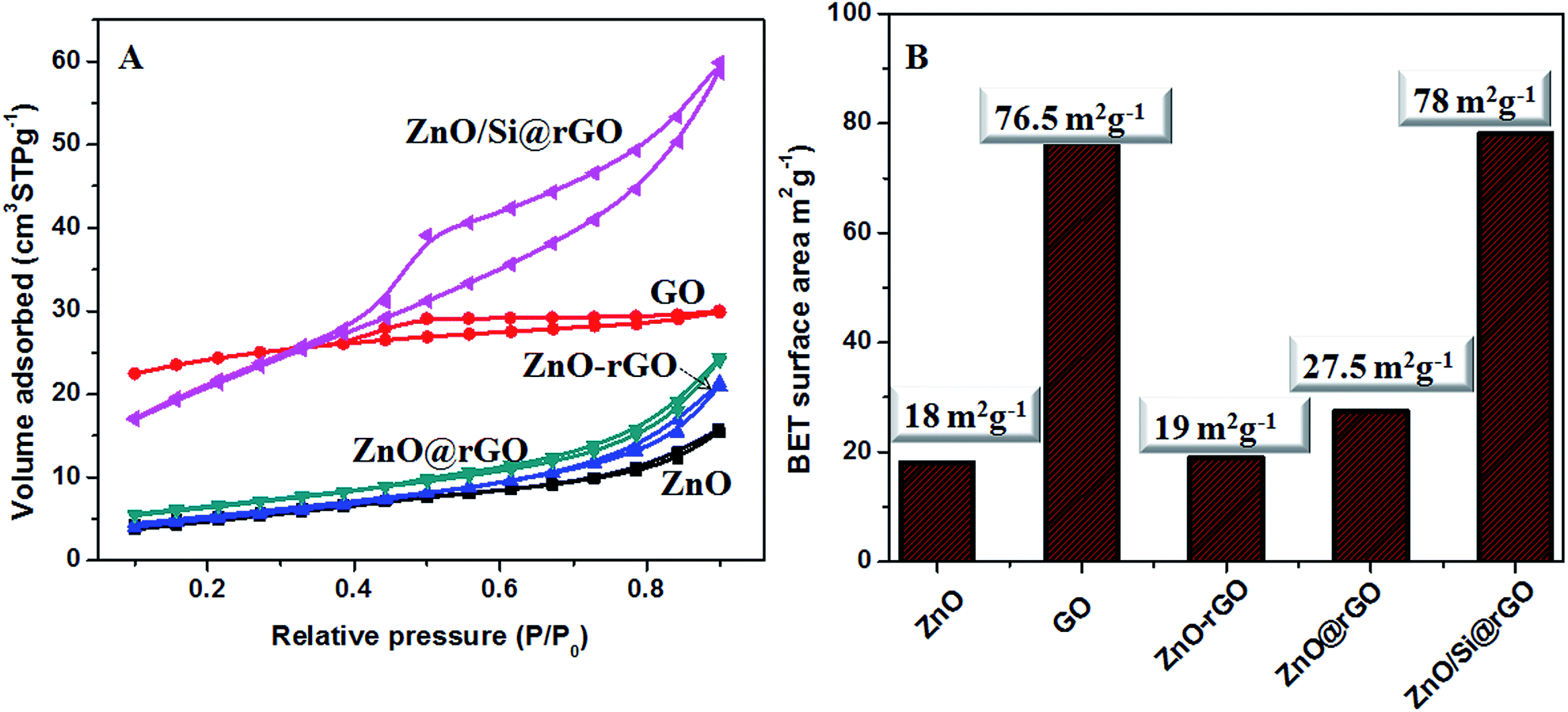 | ||
| Fig. 5 (A) N2 adsorption–desorption isotherms for ZnO, GO, ZnO–rGO, ZnO@rGO and ZnO/Si@rGO. (B) BET surface area values of ZnO, GO, ZnO–rGO, ZnO@rGO and ZnO/Si@rGO. | ||
3.3 Adsorption and photodegradation studies of organic cationic dyes
Adsorption and photoactivity of catalytically modified rGO is validated through the adsorption/degradation of a most preferred MB dye.23 The comparative performance of the ZnO and ZnO/rGO nanoarchitectures on MB dye adsorption was studied and is shown in Fig. 6A. Bare ZnO does not show any adsorption. When rGO is present, the ZnO based hybrid nanocomposite is showing ∼59% and nanocomposite is showing ∼42% of adsorption. The adsorption is mainly due to the high surface area and the mesoporous nature of rGO nanostructures. The adsorption is possibly governed by the π–π conjugation between dye molecules and the aromatic regions of the rGO as well as the ionic interactions of the dye molecules with the oxygen containing surface functional groups of the rGO nanosheets.36,37 A high BET surface area of ZnO/Si@rGO hybrid nanocomposite is a reason for a better adsorption of MB dye compared to ZnO@rGO and ZnO–rGO. | ||
| Fig. 6 (A) Adsorption studies using adsorbents ZnO, ZnO–rGO, ZnO@rGO, and ZnO/Si@rGO; (B) photoactivity studies using ZnO, ZnO–rGO, ZnO/Si@rGO and ZnO–rGO (concentration of MB = 340 μM and concentration of catalyst = 0.4 g L−1). (C) Adsorption and photodegradation study using ZnO@rGO (i) 340 μM MB (ii) after adsorption 120 min (iii) after photocatalytic activity 120 min (adsorbent dosage = 0.4 g L−1); (D) mechanism for the photodegradation of ZnO@rGO. | ||
The strong absorption in the UV region confirms the UV light activity of the samples (Fig. S3†). A comparative photocatalytic degradation of MB over ZnO and ZnO/rGO nanoarchitectures under UV light irradiation is presented in Fig. 6B. The dye degradation activity in ZnO@rGO photocatalyst is significantly high compared to the bare ZnO. About 69% of MB is degraded by ZnO@rGO within 2 h UV irradiation. This experiment shows the combined effect of ZnO and rGO for the photodegradation of MB than ZnO alone. The mechanism for the enhancement in the photodegradation of ZnO/rGO nanoarchitectures is given here.
The 2D planar conjugation structure of rGO makes this material a very good acceptor for the photogenerated electron from nano-ZnO under UV irradiation. This leads to the rapid transfer of the photogenerated electrons from the conduction band of ZnO to the rGO and helps in the suppression of the electron–hole recombination rate. This results in the enhanced photocatalytic activity of the ZnO/rGO nanoarchitectures.9 Hence the better interfacial interactions of nano-ZnO crystallites with rGO are possibly increasing the photocatalytic activity. The photocatalytic efficiency of ZnO@rGO, ZnO/Si@rGO, ZnO–rGO and ZnO is ∼69%, ∼55%, ∼49% and ∼31%, respectively. This was explained from the red shift observed in the UV-vis absorption spectra of the ZnO/rGO nanoarchitectures given in Fig. S3.† The reduced crystallite size of in situ grown ZnO in ZnO@rGO makes this material more photoactive than ZnO–rGO composite prepared via physical blending technique. Even though ZnO/Si@rGO shows good distribution of nano-ZnO over rGO sheets compared to ZnO@rGO, the presence of aminosilane crosslinking agent retards its efficiency. From the experimental data, the kapp values were calculated (Fig. S4†), and the kapp value of ZnO@rGO is 0.005 min−1, which is higher than that of bare ZnO (0.003 min−1). This high photodegradation rate constant confirms the influence of rGO in the improved photocatalytic activity of ZnO@rGO nanocomposite than bare ZnO.
In order to understand whether photocatalytic degradation is taking place under UV irradiation in the presence of ZnO/rGO nanoarchitectures, we conducted adsorption and photocatalytic degradation experiments separately by keeping the catalyst and dye solution for a time of 120 min. As shown in Fig. 6C, we could see a significant reduction in the peak intensity after 120 min of adsorption experiment, which corresponds to ∼58.12% of dye removal via surface adsorption mechanism. In the same time, ∼62.8% of dye removal was obtained after 120 min of photocatalytic experiment. It clearly indicates the influence of UV light activity of ZnO@rGO.
Fig. 7A and B show the adsorption and degradation performance of ZnO/Si@rGO and ZnO@GO on a series of cationic organic dyes (MB, MG, CV and Rh6G). ZnO/Si@rGO hybrid nanocomposites show comparatively high adsorption (∼59% of MB, ∼50% of MG, ∼28% of CV and ∼9% of Rh6G). Unfortunately, they are poor in photoactivity because the APTMS inhibits the UV interaction. In the case of ZnO@rGO, the photocatalytic activity is better (∼69% of MB, ∼67% of MG, ∼46% of CV and ∼16% of Rh6G). In both ZnO@rGO and ZnO/Si@rGO, the adsorption and degradation are showing dependence with the molecular weight of the selected cationic dyes. From Fig. 7A and B, we can understand that as the dye molecular weight increases, the adsorption and photodegradation decreases. We can see a drastic reduction in the adsorption of the dye with high molecular weight (Rh6G), which subsequently reduces the photodegradation behavior as well.
 | ||
| Fig. 7 (A) Dye adsorption (%) and (B) dye degradation (%) studies of MB, MG, CV and Rh6G using ZnO@rGO and ZnO/Si@rGO. | ||
3.4 Fabrication of functional textile filters for oil adsorption
Cotton textile substrate deposited with ZnO/Si@rGO hybrid nanocomposite was examined for the microstructures and surface wettability. The optical as well as SEM images of the cotton textile coated with ZnO/Si@rGO hybrid nanocomposites (Fig. 8F and G) showed a completely different surface texture where the cotton fibers have firmly picked the catalytic material and covered uniformly the surface with a homogeneous top-coat. In bare cotton the inter-woven fabrics are clearly visible (Fig. 8A–D), whereas in the catalytically modified rGO hybrid nanocomposites, the fabrics show a thick catalytic coating (Fig. 8E–H). | ||
| Fig. 8 (A–D) Photograph, optical image, SEM image and EDAX of bare cotton textile; (E–H) photograph, optical image, SEM image and EDAX of ZnO/Si@rGO coated cotton textile. | ||
The hydrophobic nature of the hybrid nanocomposite is demonstrated in Fig. 9B. The ZnO/Si@rGO hybrid nanocomposites particles are floating on the surface, whereas the ZnO@rGO nanocomposite is submerged fully in water medium (Fig. 9A). The hydrophobic behavior of ZnO/Si@rGO was also confirmed from the contact angle measurement. The ZnO/Si@rGO coating on cotton textile shows a contact angle of 113.7°. The contact angle of bare cotton textile is 69°. Apart from chemical modification of ZnO/rGO due to the aminosilane functional groups, the ZnO/Si@rGO also forms randomly oriented surface features depending upon fabric surface; the presence of bumps and valleys keep the water droplets at a high contact angle (Fig. 9D). In fact rGO has nano sheet layered structures in which spherically shaped ZnO nanocrystallites are grown. Since the size controlled ZnO on the flat surface of rGO create high surface roughness, the coatings show better hydrophobic property. The SEM images show the surface roughness of the ZnO/Si@rGO coating on cotton textiles (Fig. 8G). The hybrid catalyst deposited cotton textile was tested for the uptake of engine oil from water (Fig. 9E). A comparative study of oil uptake behavior of ZnO/Si@rGO coated cotton textile with bare cotton textile is given in Table 1. From the results it was found that ZnO/Si@rGO coated cotton textile shows ∼12 times greater adsorption capacity than bare cotton, and coated cotton textile can be used as photoregenerative oil sorbent.
 | ||
| Fig. 9 (A) ZnO@rGO sank into water; (B) ZnO/Si@rGO floated on water; (C) water droplet on bare cotton textile; (D) water droplet on ZnO/Si@rGO coated cotton textile; (E) ZnO/Si@rGO coated cotton textile on oil–water interface. | ||
4. Conclusions
A catalytic-adsorbent material architecture is designed with ZnO/Si@rGO hybrid nanocomposite via a facile, one-pot microwave technique. Beneficial properties of ZnO nanocrystals as well as rGO nanosheets for the combined adsorption and photodegradation were studied using different organic cationic dyes. It was found that rGO acts as dual-functional material. It imparts high surface area where in situ growth of rGO supported ZnO/Si@rGO hybrid nanocomposite produced mesoporous catalytic-sorbent with BET surface area of ∼78 m2 g−1. Moreover, the rGO favors in promoting the adsorption of cationic dyes in nano-ZnO, which otherwise fails miserably. Compared to phase pure nano-ZnO, the hybrid nanocomposite was found to retain ∼59% adsorption and ∼55% photoactivity. A thin dip-coating of mesoporous ZnO/Si@rGO nanohybrid on simple cotton textile produced a hydrophobic surface with contact angle value of 113.7°, which is receptive to selectively pick-up oil contaminants in water medium. Compared to bare cotton textile, the catalytically modified cotton textile resulted in ∼12 times more oil uptake. The uniform anchoring of size controlled nano ZnO on rGO surface via in situ addition of APTMS produced photocatalytically regenerative oil and dye adsorbing bio-filters.Acknowledgements
The authors are gratefully thanking the Director, CSIR-National Institute of Science and Technology (NIIST) for providing the laboratory facilities. K. B. Babitha is grateful to CSIR, India, for providing the Senior Research Fellowship. Authors thank Dr Prabhakar Rao, Mr Kiran Mohan, Mrs Lucy Paul and Mr Prakash S. P. for XRD, TEM, SEM and FTIR analysis, respectively. All the members of the Materials Science and Technology Division are acknowledged for providing general support.References
- X. An and J. C. Yu, RSC Adv., 2011, 1, 1426–1434 RSC.
- W. Tu, Y. Zhou and Z. Zou, Adv. Funct. Mater., 2013, 23, 4996–5008 CrossRef CAS PubMed.
- Y. W. Wang, A. Cao, Y. Jiang, X. Zhang, J. H. Liu, Y. Liu and H. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 2791–2798 CAS.
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC.
- R. K. Upadhyay, N. Soinb and S. S. Roy, RSC Adv., 2014, 4, 3823–3851 RSC.
- J. Wang, T. Tsuzuki, B. Tang, X. Hou, L. Sun and X. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 3084–3090 CAS.
- Q. Zhang, C. Tian, A. Wu, T. Tan, L. Sun, L. Wang and H. Fu, J. Mater. Chem., 2012, 22, 11778–11784 RSC.
- X. Yang, C. Chen, J. Li, G. Zhao, X. Ren and X. Wang, RSC Adv., 2012, 2, 8821–8826 RSC.
- L. Gu, J. Wang, H. Cheng, Y. Zhao, L. Liu and X. Han, ACS Appl. Mater. Interfaces, 2013, 5, 3085–3093 CAS.
- L. Jia, D. H. Wang, Y. X. Huang, A. W. Xu and H. Q. Yu, J. Phys. Chem. C, 2011, 115, 11466–11473 CAS.
- N. Zhang, Y. Zhang, X. Pan, M. Q. Yang and Y. J. Xu, J. Phys. Chem. C, 2012, 116, 18023–18031 CAS.
- V. Stengl, D. Popelkov and P. Vlacil, J. Phys. Chem. C, 2011, 115, 25209–25218 CAS.
- X. Liu, L. Pan, T. Lv, T. Lu, G. Zhu, Z. Suna and C. Sunb, Catal. Sci. Technol., 2011, 1, 1189–1193 CAS.
- M. B. M. Krishna, N. Venkatramaiah, R. Venkatesanb and D. N. Rao, J. Mater. Chem., 2012, 22, 3059–3068 RSC.
- J. Manna, G. Begum, K. P. Kumar, S. Misra and R. K. Rana, ACS Appl. Mater. Interfaces, 2013, 5, 4457–4463 CAS.
- K. Jayanthi, S. Chawla, A. G. Joshi, Z. H. Khan and R. K. Kotnala, J. Phys. Chem. C, 2010, 114, 18429–18434 CAS.
- L. W. Sun, H. Q. Shi, W. N. Li, H. M. Xiao, S. Y. Fu, X. Z. Cao and Z. X. Li, J. Mater. Chem., 2012, 22, 8221–8227 RSC.
- P. Hareesh, K. B. Babitha and S. Shukla, J. Hazard. Mater., 2012, 229, 177–182 CrossRef PubMed.
- K. V. Baiju, S. Shukla, K. S. Sandhya, J. James and K. G. K. Warrier, J. Phys. Chem. C, 2007, 111, 7612–7622 CAS.
- K. B. Jaimy, K. V. Baiju, S. K. Ghosh and K. G. K. Warrier, J. Solid State Chem., 2012, 186, 149–157 CrossRef CAS PubMed.
- P. S. Suchithra, V. Linsha, A. Peer Mohamed and S. Ananthakumar, J. Chem. Eng., 2012, 200, 589–600 CrossRef PubMed.
- Y. Zhao, X. Song, Q. Song and Z. Yin, CrystEngComm, 2012, 14, 6710–6719 RSC.
- W. S. Wang, D. H. Wang, W. G. Qu, L. Q. Lu and A. W. Xu, J. Phys. Chem. C, 2012, 116, 19893–19901 CAS.
- R. Liao, Z. Tang, T. Lin and B. Guo, ACS Appl. Mater. Interfaces, 2013, 5, 2174–2181 CAS.
- S. Gawande and S. R. Thakare, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2013, 52, 614–618 Search PubMed.
- X. F. Wu, J. Zhang, Y. F. Zhuang, S. D. Fu and X. Y. Yu, Micro Nano Lett., 2014, 9, 804–806 Search PubMed.
- H. Q. Shi, W. N. Li, L. W. Sun, Y. Liu, H. M. Xiao and S. Y. Fu, Chem. Commun., 2011, 47, 11921–11923 RSC.
- D. Costenaro, F. Carniato, G. Gatti, C. Bisio and L. Marchese, J. Phys. Chem. C, 2011, 115, 25257–25265 CAS.
- M. Long, Y. Qin, C. Chen, X. Guo, B. Tan and W. Cai, J. Phys. Chem. C, 2013, 117, 16734–16741 CAS.
- L. Valentini, S. B. Bon, O. Monticellib and J. M. Kennya, J. Mater. Chem., 2012, 22, 6213–6217 RSC.
- A. Danon, P. C. Stair and E. Weitz, J. Phys. Chem. C, 2011, 115, 11540–11549 CAS.
- S. Mallakpour and M. Iderli, Polym. Bull., 2013, 70, 2137–2149 CrossRef CAS.
- C. Bressy, V. G. Ngo, F. Ziarelli and A. Margaillan, Langmuir, 2012, 28, 3290–3297 CrossRef CAS PubMed.
- Y. Guo, H. Wang, C. He, L. Qiu and X. Cao, Langmuir, 2009, 8, 4678–4684 CrossRef PubMed.
- R. Y. Hong, J. H. Li, L. L. Chen, D. Q. Liu, H. Z. Li, Y. Zheng and J. Ding, Powder Technol., 2009, 189, 426–432 CrossRef CAS PubMed.
- P. Sharma and M. R. Das, J. Chem. Eng. Data, 2013, 58, 151–158 CrossRef CAS.
- J. Xu, L. Wang and Y. Zhu, Langmuir, 2012, 28, 8418–8425 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: TEM and EDX spectra of ZnO@rGO and ZnO/Si@rGO, TEM and the corresponding SAED pattern of ZnO–rGO composite, UV-vis spectra and the plot of ln(C0/C) versus t for ZnO and ZnO/rGO nanoarchitectures. See DOI: 10.1039/c5ra04850h |
| This journal is © The Royal Society of Chemistry 2015 |