
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Colour tuning by the ring roundabout: [Ir(C^N)2(N^N)]+ emitters with sulfonyl-substituted cyclometallating ligands†
Cathrin D.
Ertl
a,
Jesús
Cerdá
b,
José M.
Junquera-Hernández
b,
Antonio
Pertegás
b,
Henk J.
Bolink
b,
Edwin C.
Constable
a,
Markus
Neuburger
a,
Enrique
Ortí
*b and
Catherine E.
Housecroft
*a
aDepartment of Chemistry, University of Basel, Spitalstrasse 51, CH-4056 Basel, Switzerland. E-mail: catherine.housecroft@unibas.ch
bInstituto de Ciencia Molecular, Universidad de Valencia, 46980-Paterna, Valencia, Spain. E-mail: enrique.orti@uv.es
First published on 13th May 2015
Abstract
A series of cationic bis-cyclometallated iridium(III) complexes [Ir(C^N)2(N^N)]+ is reported. Cyclometallating C^N ligands are based on 2-phenylpyridine with electron-withdrawing sulfone substituents in the phenyl ring: 2-(4-methylsulfonylphenyl)pyridine (H1) and 2-(3-methylsulfonylphenyl)pyridine (H2). 2-(1H-Pyrazol-1-yl)pyridine (pzpy) and 2-(3,5-dimethyl-1H-pyrazol-1-yl)pyridine (dmpzpy) are used as electron-rich ancillary N^N ligands. The complexes have been fully characterized and the single crystal structure of [Ir(2)2(dmpzpy)][PF6]·MeCN has been determined. Depending on the position of the methylsulfonyl group, the complexes are green or blue emitters with vibrationally structured emission maxima at 491, 523 nm for [Ir(1)2(N^N)][PF6] or 463, 493 nm for [Ir(2)2(N^N)][PF6] in MeCN solution. The marked vibrational structure and the absence of a rigidochromic shift, together with theoretical predictions based on density functional theory calculations, confirm the 3LC nature of the emitting triplet state. All four complexes have relatively high photoluminescence quantum yields in de-aerated solution (53 to 77%). On going from solution to powder samples, the emission is red-shifted and the quantum yields are considerably lower (≤11%). The complexes were tested in light-emitting electrochemical cells (LECs) achieving maximum luminances of 141 cd m−2 when operated at 100 A m−2 using pulsed current driving conditions.
Introduction
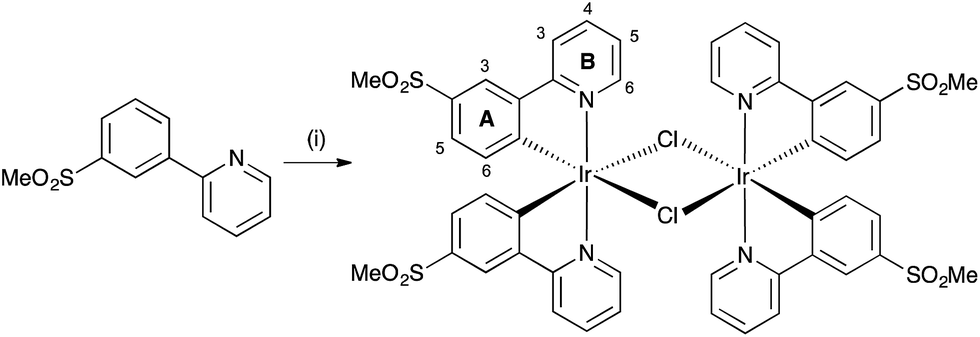
Cationic iridium(III) complexes of the type [Ir(C^N)2(N^N)]+, in which C^N is a cyclometallated ligand and N^N is a diimine ligand, are well-established emissive materials in light-emitting electrochemical cells (LECs).1,2 Prototype LECs incorporated conjugated polymers in the active layer,3 but the development of LECs with emissive ionic transition-metal complexes (iTMCs) has led to single-component devices.1 The application of a bias across the device causes the ions in the active layer to migrate towards the electrodes where their accumulation results in a drop in potential near the electrode surface. The formation of ionic junctions lowers the barrier for electron and hole injection and, as a consequence, the device is independent of the work function of the electrode material.4,5 Hence, it is not a prerequisite that LECs use low work-function metals, as is necessary in organic light-emitting diodes (OLEDs), and air-stable electrodes (e.g. Al) are employed in LECs. This has a beneficial consequence: the rigorous exclusion of air, which is essential in the fabrication of OLEDs, is unnecessary in LECs. Another advantage of LECs over OLEDs is their far simpler architecture and the fact that they can be assembled using solution processing.1Despite the fabrication advantages of LECs, there is a lack of wide bandgap iTMCs to produce the blue iTMC-LECs needed to combine with orange emitters for white light. In [Ir(C^N)2(N^N)]+ complexes, the HOMO contains contributions from the Ir(III) dπ orbitals and phenyl π-orbitals of the cyclometallated C^N ligands, and the LUMO is usually localized on the N^N ligand.1 The localized character of the HOMO–LUMO manifold lends itself perfectly to colour tuning the emission of [Ir(C^N)2(N^N)]+ complexes by introducing N^N ligands that destabilize the LUMO and/or C^N ligands that stabilize the HOMO. In 2006, Nazeeruddin et al. showed that [Ir(ppy)2(4,4′-(Me2N)2bpy)]+ (Hppy = 2-phenylpyridine, 4,4′-(Me2N)2bpy = 4,4′-bis(dimethylamino)-2,2′-bipyridine) was an efficient blue-green emitter,6 and pointed to the potential use of 1-phenylpyrazole or fluorinated 2-phenylpyridine ligands to stabilize the HOMO of [Ir(C^N)2(N^N)]+. This approach to blue emission was demonstrated in 2005 by Thompson and coworkers in a landmark paper.7 The quest for blue emitters8–21 continues, with utilization of the structure–property ‘rules’, leading to the replacement of 1-phenylpyrazolyl- by 2-methyl-5-phenyl-2H-tetrazolyl-functionalized C^N ligands.22 The number and position of fluoro groups in Hppy have only a small influence on the photophysical properties of [Ir(C^N)2(4,4′-tBu2bpy)]+ complexes (4,4′-tBu2bpy = 4,4′-di-tert-butyl-2,2′-bipyridine),23 although increasing the number of fluorine atoms leads to shorter-lived LECs.23 Indeed, the use of fluorinated C^N ligands in iridium(III) complexes in OLEDs has been questioned due to the instability of the Cphenyl–F bond;24 hence, fluorine-free blue [Ir(C^N)2(N^N)]+ emitters are desirable. It must be mentioned that blue photoluminescence (PL) in solution, thin-film or powder, is no guarantee of blue electroluminescence (EL) under device conditions, and significant shifts in emission maxima on going from PL to EL are common.25
Following studies of blue emitting complexes for OLEDs,26,27 Evariste et al.28 demonstrated the use of cyclometallated methyl- and methoxy-substituted 2,3′-bipyridine ligands as the C^N domain in blue-emitting [Ir(C^N)2(4,4′-tBu2bpy)]+ complexes. An alternative approach is to replace the fluoro-substituents in the C^N ligands by electron-withdrawing sulfone groups, a design strategy previously demonstrated in OLEDs,29 and one that we have applied in [Ir(C^N)2(N^N)]+ complexes (Scheme 1).30,31 However, the complexes in Scheme 1 are green emitters and we consider here further modification to achieve a shift in emission to higher energies. An appealing strategy is the use of sulfone-functionalized C^N ligands and derivatives of 1-phenylpyrazole as the N^N ligand, combining the effects of the electron-withdrawing sulfone groups with those of the electron-rich pyrazole ring, leading to larger HOMO–LUMO gaps in [Ir(C^N)2(N^N)]+ complexes. Stereoisomerism in [Ir(C^N)3] emitters in OLEDs is important for the enhancement of phosphoresence quantum yield and electrochemical stability, with the fac-isomer being superior to the mer-isomer,32 and we have now investigated the effect of positional isomerization in the sulfone-functionalized ligands as it is known that the emission behaviour of some iridium complexes33–35 is sensitive to the positions of the substituents in the C^N ligands. Density functional theory (DFT) calculations have shown that photophysical properties of cationic [Ir(ppy)2(P^P)]+ complexes vary with the position of functionalization of the [ppy]− ligands.36
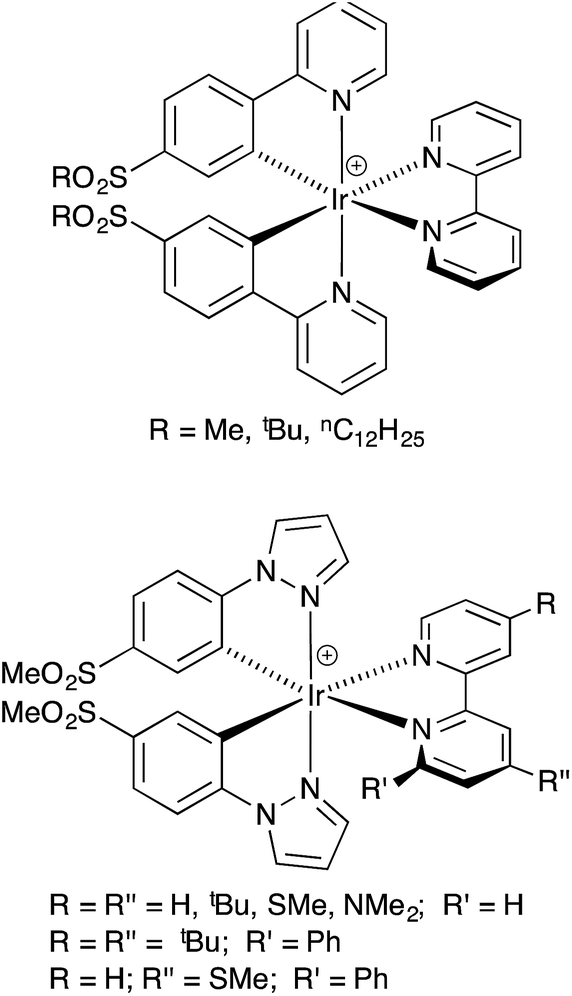 | ||
| Scheme 1 Previously reported [Ir(C^N)2(N^N)]+ complexes containing sulfone-functionalized C^N ligands.30,31 | ||
To further blue-shift the emission, we have synthesized a series of complexes combining sulfone-substituted H1 and H2 and pyrazolylpyridine ancillary ligands (Scheme 2). The complex [Ir(ppy)2(pzpy)][PF6] previously prepared by He et al.8 is used as a reference compound for the comparison of the photophysical and electrochemical properties of the new complexes (Scheme 3), which are further analysed through DFT calculations. The electroluminescent properties of the complexes have been investigated in LEC devices operated under pulsed driving conditions.
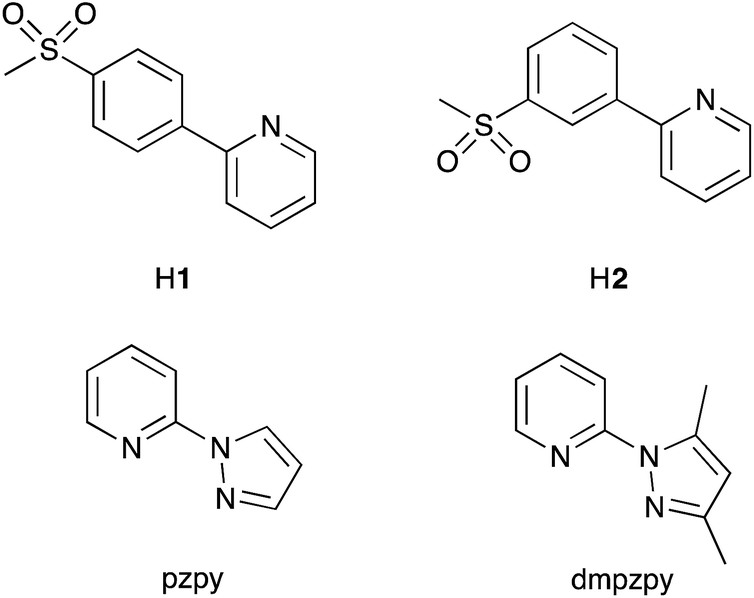 | ||
| Scheme 2 Structures of cyclometallating ligands 2-(4-methylsulfonylphenyl)pyridine (H1) and 2-(3-methylsulfonylphenyl)pyridine (H2) and ancillary ligands 2-(1H-pyrazol-1-yl)pyridine (pzpy) and 2-(3,5-dimethyl-1H-pyrazol-1-yl)pyridine (dmpzpy). | ||
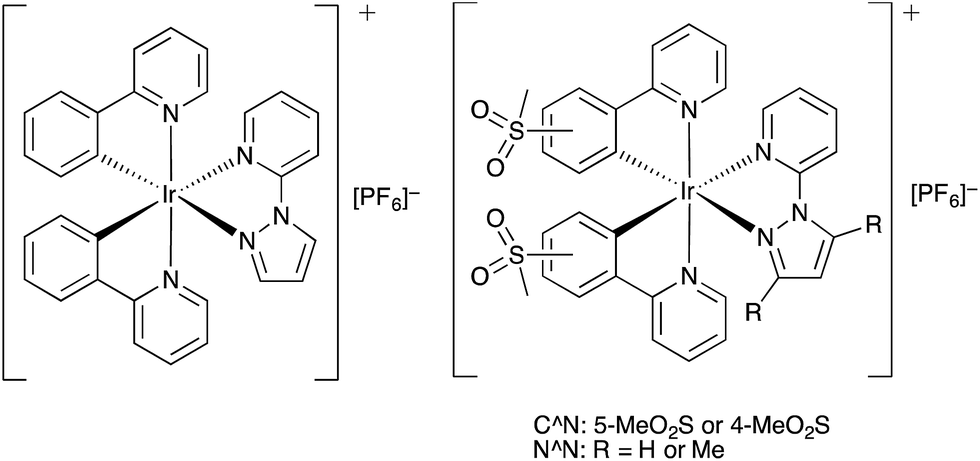 | ||
| Scheme 3 Structures of [Ir(ppy)2(pzpy)][PF6]8 and methylsulfonyl-substituted complexes [Ir(C^N)2(N^N)][PF6] with C^N = [1]− or [2]−, and N^N = 2-(1H-pyrazol-1-yl)pyridine (pzpy) or 2-(3,5-dimethyl-1H-pyrazol-1-yl)pyridine (dmpzpy). | ||
Experimental
General experimental, ligand synthesis and IR spectroscopic data for the complexes are given in the ESI.†Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), 280 sh (39000), 420 sh (3000). Emission (CH3CN, 1.0 × 10−5 mol dm−3, λexc = 261 nm) λmaxem = 491, 523 nm. ESI-MS m/z 802.4 [M-PF6]+ (calc. 802.1). Found C 40.24, H 3.28, N 7.51; C32H27F6IrN5O4PS2·0.5H2O requires C 40.21, H 2.95, N 7.33%.
000), 285 sh (38000), 420 sh (2900). Emission (CH3CN, 1.0 × 10−5 mol dm−3, λexc = 261 nm) λmaxem = 491, 523 nm. ESI-MS m/z 830.5 [M-PF6]+ (calc. 830.1). Found C 42.25, H 3.51, N 7.47; C34H31F6IrN5O4PS2 requires C 41.89, H 3.20, N 7.18%.
000), 280 sh (44000), 375 sh (5000). Emission (CH3CN, 1.0 × 10−5 mol dm−3, λexc = 259 nm) λmaxem = 463, 493 nm. ESI-MS m/z 802.5 [M–PF6]+ (calc. 802.1). Found C 39.62, H 3.20, N 7.51; C32H27F6IrN5O4PS2·H2O requires C 39.83, H 3.03, N 7.26%.
000), 280 sh (41000), 375 sh (4300). Emission (CH3CN, 1.0 × 10−5 mol dm−3, λexc = 259 nm) λmaxem = 463, 493 nm. ESI-MS m/z 830.5 [M-PF6]+ (calc. 830.1). Found C 41.89, H 3.54, N 7.22; C34H31F6IrN5O4PS2 requires C 41.89, H 3.20, N 7.18%.
000), 280 sh (39000), 420 sh (3000). Emission (CH3CN, 1.0 × 10−5 mol dm−3, λexc = 261 nm) λmaxem = 491, 523 nm. ESI-MS m/z 802.4 [M-PF6]+ (calc. 802.1). Found C 40.24, H 3.28, N 7.51; C32H27F6IrN5O4PS2·0.5H2O requires C 40.21, H 2.95, N 7.33%.
000), 285 sh (38000), 420 sh (2900). Emission (CH3CN, 1.0 × 10−5 mol dm−3, λexc = 261 nm) λmaxem = 491, 523 nm. ESI-MS m/z 830.5 [M-PF6]+ (calc. 830.1). Found C 42.25, H 3.51, N 7.47; C34H31F6IrN5O4PS2 requires C 41.89, H 3.20, N 7.18%.
000), 280 sh (44000), 375 sh (5000). Emission (CH3CN, 1.0 × 10−5 mol dm−3, λexc = 259 nm) λmaxem = 463, 493 nm. ESI-MS m/z 802.5 [M–PF6]+ (calc. 802.1). Found C 39.62, H 3.20, N 7.51; C32H27F6IrN5O4PS2·H2O requires C 39.83, H 3.03, N 7.26%.
000), 280 sh (41000), 375 sh (4300). Emission (CH3CN, 1.0 × 10−5 mol dm−3, λexc = 259 nm) λmaxem = 463, 493 nm. ESI-MS m/z 830.5 [M-PF6]+ (calc. 830.1). Found C 41.89, H 3.54, N 7.22; C34H31F6IrN5O4PS2 requires C 41.89, H 3.20, N 7.18%.
Crystallography
Single crystal data were collected on a Bruker APEX-II diffractometer; data reduction, solution and refinement used APEX.37 Structure analysis used Mercury v. 3.5.1.38748 reflections, 6985 unique, Rint = 0.049. Refinement of 6361 reflections (505 parameters) with I > 2σ (I) converged at final R1 = 0.0239 (R1 all data = 0.0251), wR2 = 0.0257 (wR2 all data = 0.0282), gof = 1.0886.
Computational details
DFT calculations were carried out with the D.01 revision of the Gaussian 09 program package39 using Becke's three-parameter B3LYP exchange-correlation functional40,41 together with the 6-31G** basis set for C, H, N, S and O42 and the “double-ζ” quality LANL2DZ basis set for Ir.43 The geometries of the singlet ground state (S0) and of the lowest-energy triplet state (T1) were fully optimized without imposing any symmetry restriction. The geometry of T1 was calculated at the spin-unrestricted UB3LYP level with a spin multiplicity of three. Phosphorescence emission energies were estimated as the vertical difference between the energy of the minimum of the lowest-energy triplet state and the energy of S0 at the T1 optimized geometry. All the calculations were performed in the presence of the solvent (acetonitrile). Solvent effects were considered within the self-consistent reaction field (SCRF) theory using the polarized continuum model (PCM) approach.44–46 The calculation of the energy of S0 at the T1 geometry was performed as an equilibrium single-point calculation with respect to the solvent reaction field/solute electronic density polarization process. Time-dependent DFT (TD-DFT)47–49 calculations of the lowest lying 15 triplets were performed in the presence of the solvent at the minimum-energy geometry optimized for the ground state.Device preparation and characterization
Electroluminescent LEC devices were made as follows. First, glass substrates with sputtered ITO contact (Naranjo Substrates) were cleaned by sequentially washing and sonication with soap, deionized water, isopropanol and a UV-O3 lamp for 20 min. Then, an 80 nm layer of poly(3,4-ethylenedioxythiophene):polystyrenesulfonate (PEDOT:PSS) (CLEVIOS™ P VP AI 4083, aqueous dispersion, 1.3–1.7% solid content, Heraeus) was spin-coated on the ITO substrates to improve the reproducibility of the devices and to prevent the formation of pinholes. After this layer deposition, a 100 nm transparent film of the iTMC and the ionic liquid (IL) 1-butyl-3-methylimidazolium hexafluoridophosphate ([Bmim][PF6]) (>98.5%, Sigma-Aldrich) in a 4:1 molar ratio were spin-coated from a 20 mg mL−1 acetonitrile solution. The devices were transferred into an inert atmosphere glovebox (<0.1 ppm O2 and H2O, M. Braun). The Al electrode (70 nm) was thermally vapour-deposited using a shadow mask under a vacuum (<1 × 10−6 mbar) with an Edwards Auto500 evaporator integrated in the glovebox. The final device configuration was ITO/PEDOT:PSS/iTMC:IL/Al. The device characteristics were measured by applying a pulsed current (average current density 100 A m−2, 50% duty cycle, 1 kHz, block wave) and monitoring the current and luminance versus time by a True Colour Sensor MAZeT (MTCSiCT Sensor) with a Botest OLT OLED Lifetime-Test System. The electroluminescent spectra were measured using an Avantes AvaSpec-2048 Fiber Optic Spectrometer.
Results and discussion
Ligand synthesis and characterization
The sulfone-substituted cyclometallating ligand H2 was prepared in three steps in an analogous manner to the synthesis of 2-(4-methylsulfonyl)phenylpyridine (H1),31 starting from 2-bromopyridine and 3-fluorophenylboronic acid. The first step was a Suzuki coupling in EtOH/H2O with PdCl2,50 followed by a nucleophilic aromatic substitution with NaSMe to yield the corresponding thioether derivative. Oxidation of sulfur using H2O2/Na2WO4 in MeOH to give H2 (Scheme 4) was performed under mild conditions. High yields were achieved in all three steps. Compound H2 was characterized using multinuclear NMR spectroscopy, mass spectrometry, infrared spectroscopy and elemental analysis. The base peak in the LC-ESI mass spectrum corresponded to [M + H]+ with an isotope pattern matching that calculated. 1H and 13C NMR signals were assigned using COSY, HMQC and HMBC methods. Pyrazolylpyridine ancillary ligands were synthesized according to literature.51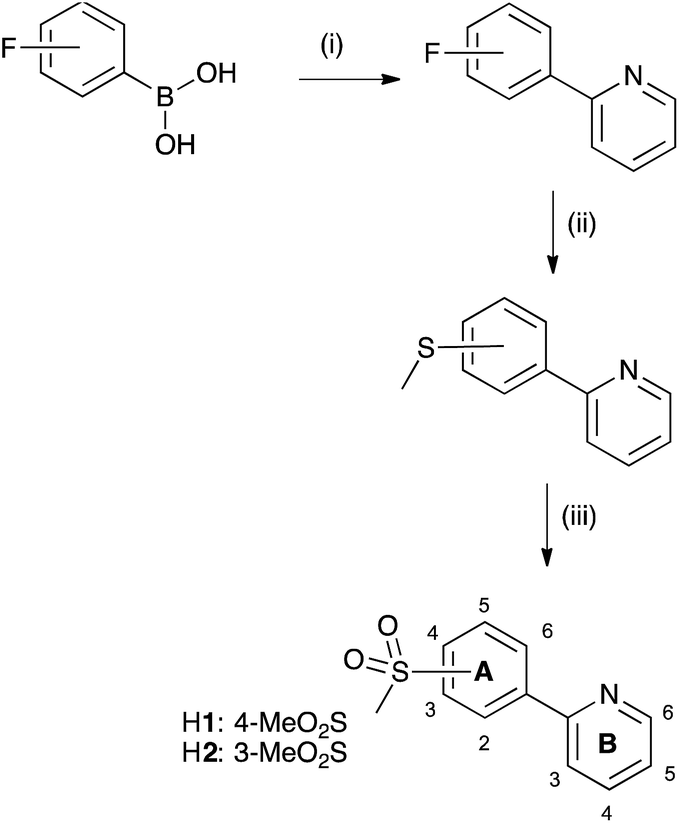 | ||
| Scheme 4 Synthetic route to H1 and H2. Conditions: (i) 2-bromopyridine, PdCl2, K2CO3, EtOH/H2O 1:1, overnight, room temperature or reflux; (ii) NaSMe, N-methyl-2-pyrrolidone, N2, microwave reactor, 1 h, 80 °C or 120 °C; (iii) H2O2, Na2WO4·2H2O, MeOH, overnight, room temperature. Numbering scheme is for NMR spectroscopic assignments. | ||
Synthesis and characterization of [Ir(C^N)2Cl]2 dimers
Iridium complexes [Ir(C^N)2(N^N)]+ are usually synthesized by cleavage of the chlorido-bridged iridium dimer with the corresponding ancillary ligand. These dimers are typically obtained by reaction of IrCl3·xH2O and the cyclometallating ligand under reflux conditions.52 However, for [Ir(C^N)2Cl]2 dimers containing sulfone-substituted cyclometallating ligands, we have found that a route starting from [Ir(cod)Cl]253 (cod = cycloocta-1,5-diene) is more convenient (Scheme 5), and [Ir(2)2Cl]2 was therefore prepared in an analogous manner to [Ir(1)2Cl]2.31 The dimers were isolated in high yields as yellow solids. [Ir(2)2Cl]2 proved to be poorly soluble in common organic solvents but could be characterized by 1H NMR spectroscopy. In the LC-ESI mass spectrum of a MeOH solution of the dimer, the base peak at m/z 657.1 was assigned to the [Ir(2)2]+ ion. Further intense peaks were observed at m/z 698.1 and 739.1 and corresponded to [Ir(2)2(MeCN)]+ and [Ir(2)2(MeCN)2]+, respectively; the MeCN arises from the eluent in the LC column.
 | ||
| Scheme 5 Synthetic route to [Ir(2)2Cl]2. Conditions: (i) [Ir(cod)Cl]2, 2-ethoxyethanol, N2, overnight, reflux. Numbering scheme is for NMR spectroscopic assignments. | ||
Synthesis and characterization of [Ir(C^N)2(N^N)][PF6] complexes
Complexes [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) were synthesized from the corresponding dimers and ancillary ligands pzpy and dmpzpy in MeOH in a microwave reactor (Scheme 6). Subsequent anion exchange yielded the products as yellow powders in good yields.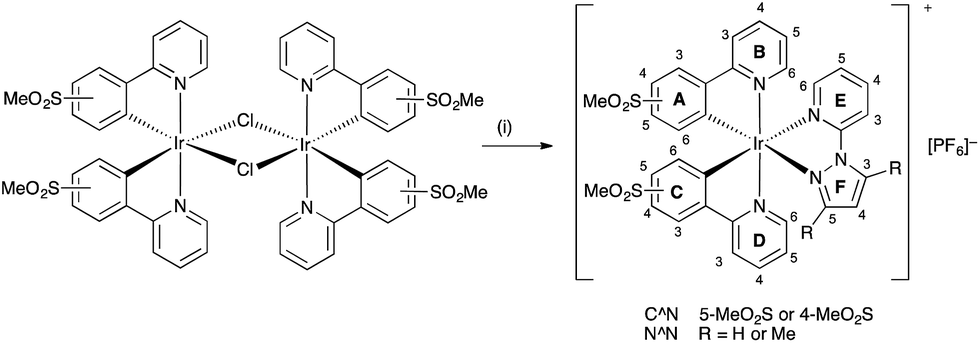 | ||
| Scheme 6 Synthetic route to [Ir(C^N)2(N^N)][PF6] with C^N = [1]− and [2]− and N^N = 2-(1H-pyrazol-1-yl)pyridine (pzpy) or 2-(3,5-dimethyl-1H-pyrazol-1-yl)pyridine (dmpzpy). Conditions: (i) pzpy or dmpzpy, MeOH, microwave reactor, 1 h, 120 °C; NH4PF6. Numbering scheme is for NMR spectroscopic assignments. | ||
In the ESI mass spectra of [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy), the base peak in each case corresponded to [M–PF6]+ and exhibited a characteristic iridium isotope pattern. 1H and 13C NMR spectra were assigned using COSY, NOESY, HMQC and HMBC methods and were consistent with the structures shown in Scheme 6. The 1H NMR spectrum of complex [Ir(2)2(pzpy)][PF6] is shown in Fig. 1 as a representative example. The cyclometallating ligands are inequivalent due to the asymmetric pzpy ligand; the most characteristic signals are those for HA6 and HC6 which occur at lowest frequency. The inequivalent cyclometallating ligands were distinguished by NOESY cross peaks between protons HE6 and HA6, and between protons HF5/HF5-Me and HB6 and/or HC6 (see Scheme 6 for numbering).
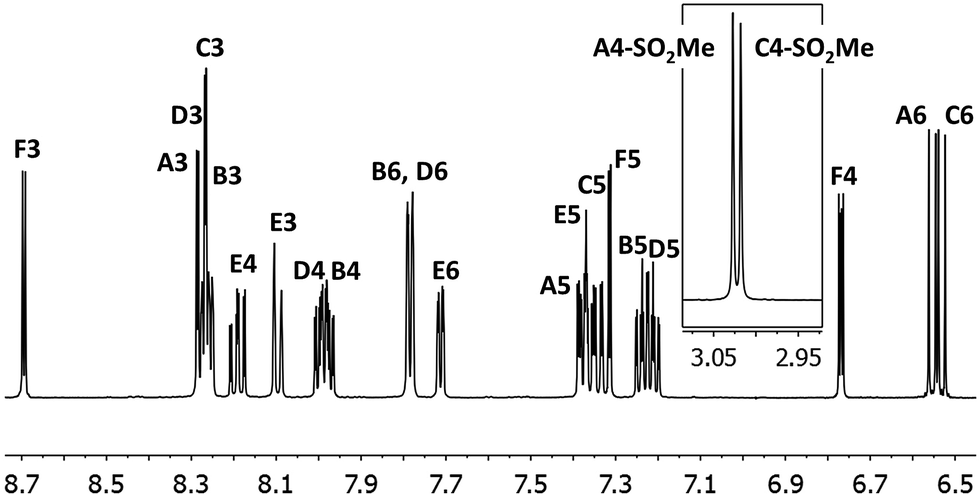 | ||
| Fig. 1 500 MHz 1H NMR spectrum of complex [Ir(2)2(pzpy)][PF6] in CD3CN with signal assignments. See Scheme 6 for proton labelling. Scale: δ/ppm. | ||
Crystal structure of [Ir(2)2(dmpzpy)][PF6]·MeCN
X-Ray quality crystals of [Ir(2)2(dmpzpy)][PF6]·MeCN were grown by overlaying a MeCN solution with Et2O, and structural determination confirmed the expected octahedral environment of the iridium(III) centre and the presence of three chelating ligands. The cation is necessarily chiral and the asymmetric unit contains the Λ-[Ir(2)2(dmpzpy)]+ cation shown in Fig. 2; important bond parameters are given in the figure caption. The compound crystallizes in the monoclinic space group C2/c and the unit cell therefore contains both enantiomers. Both the [PF6]− anion and MeCN solvent molecule are ordered. Analyses of the structures of aryl–alkyl sulfones54 and diaryl sulfones54,55 demonstrate the presence of intra- and intermolecular CHaryl⋯OS hydrogen bonds, and we have observed this feature both in the structures of the ligand H1 and the iridium dimer [Ir(1)2Cl]2.31 In the [Ir(2)2(dmpzpy)]+ cation, the sulfone groups are twisted with respect to the phenyl rings to which they are bonded with torsion angles of O1–S1–C18–C17 = 27.4(3), O2–S1–C18–C19 = −22.4(3), O3–S2–C30–C31 = 15.0(3) and O4–S2–C30–C29 = −35.1(2)° giving rise to short Osulfone⋯HCphenyl contacts in the range 2.6–2.7 Å. Intermolecular Osulfone⋯HCarene and Osulfone⋯HCmethyl interactions are dominant packing interactions along with PFanion⋯HC contacts. The [Ir(2)2(dmpzpy)]+ cations pack into sheets that are coparallel with the bc-plane (Fig. 3).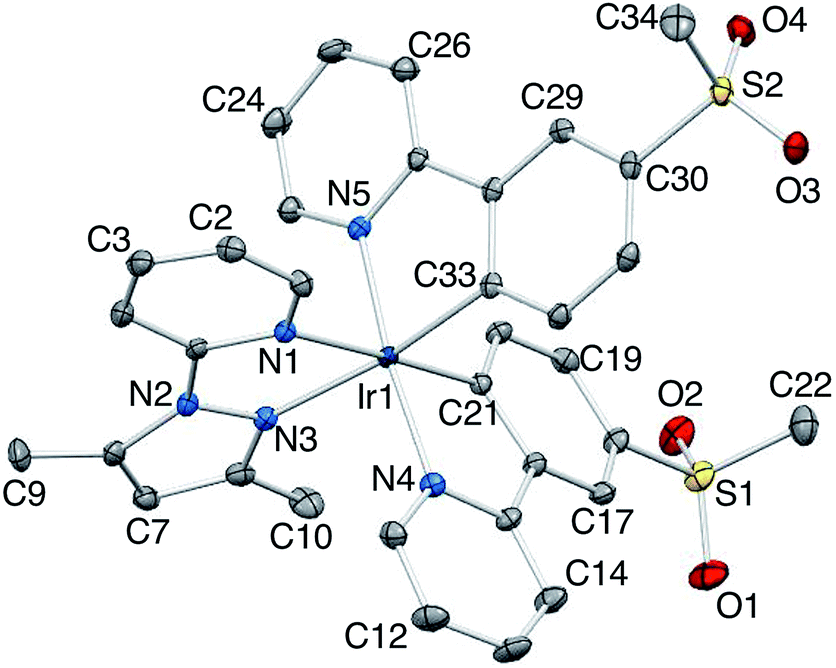 | ||
| Fig. 2 Structure of the Λ-[Ir(2)2(dmpzpy)]+ cation in [Ir(2)2(dmpzpy)][PF6]·MeCN; ellipsoids plotted at 40% probability level and H atoms omitted for clarity. Selected bond lengths and angles: Ir1–N1 = 2.144(2), Ir1–N3 = 2.126(2), Ir1–N4 = 2.050(2), Ir1–N5 = 2.044(2), Ir1–C21 = 1.998(2), Ir1–C33 = 2.012(3), C34–S2 = 1.763(3), S1–O1 = 1.435(2), S1–O2 = 1.441(2), S2–O3 = 1.442(2), S2–O4 = 1.437(2), C22–S1 = 1.769(3), N2–N3 = 1.385(3) Å; N1–Ir1–N3 = 75.53(8), N4–Ir1–C21 = 80.34(9), N5–Ir1–C33 = 80.67(9), N4–Ir1–N5 = 172.73(8), N1–Ir1–C21 = 178.07(9), N3–Ir1–C33 = 171.26(9), O1–S1–O2 = 119.32(14), O3–S2–O4 = 118.32(12)°. | ||
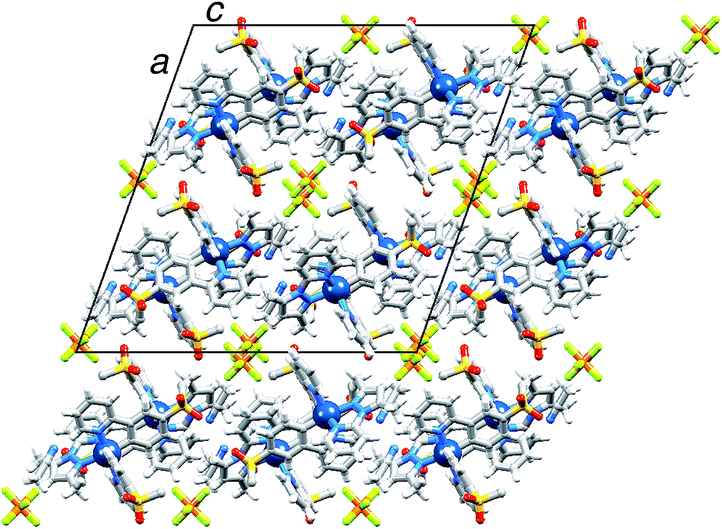 | ||
| Fig. 3 Packing diagram for [Ir(2)2(dmpzpy)][PF6]·MeCN showing sheets of cations in the bc-plane separated by [PF6]− anions. | ||
A preliminary structure of [Ir(1)2(dmpzpy)][PF6] was determined with crystals grown from a MeCN solution of the complex overlaid with Et2O. This revealed the octahedral metal centre as well as the presence of the three bidentate ligands, confirming the expected structure of the complex.
Photophysical properties
Absorption spectra of [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) were recorded in MeCN solutions (Fig. 4). The spectra are similar and the most intense absorption bands lie in the UV region and are assigned to spin-allowed, ligand-centred π* ← π transitions. Between 350 and 450 nm, less intense bands arise from 1MLCT and 1LLCT transitions.1 The lower energy absorption tails of [Ir(1)2(N^N)][PF6] extend further into the visible than those of [Ir(2)2(N^N)][PF6], with broad, but weak, maxima at 390 and 420 nm (Fig. 4).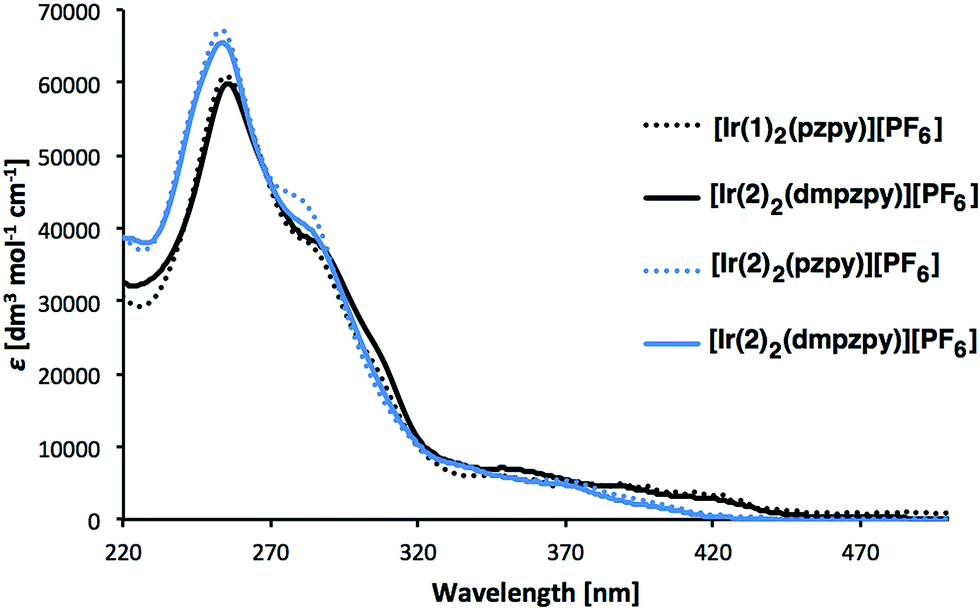 | ||
| Fig. 4 Absorption spectra of 1 × 10−5 M MeCN solutions of complexes [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy). | ||
The photoluminescence spectra of the complexes were recorded in MeCN solution both at room temperature and at 77 K and are shown in Fig. 5; emission maxima are summarized in Table 1. The emission profile is independent of the excitation wavelength. In complexes of the type [Ir(C^N)2(N^N)]+, emission occurs from the lowest-lying triplet state (T1), which comprises contributions from metal-to-ligand/ligand-to-ligand charge-transfer 3MLCT/3LLCT and ligand-centred 3LC states. In general, the higher the charge-transfer character, the broader and less structured the emission profile. The vibrational structure observed in Fig. 5a for the spectra at room temperature indicates a large 3LC character of the emissive state. The spectra at 77 K confirm this assignment because the vibrational peaks preserve their position showing almost no rigidochromic shift with respect to room temperature (Fig. 5b and Table 1).
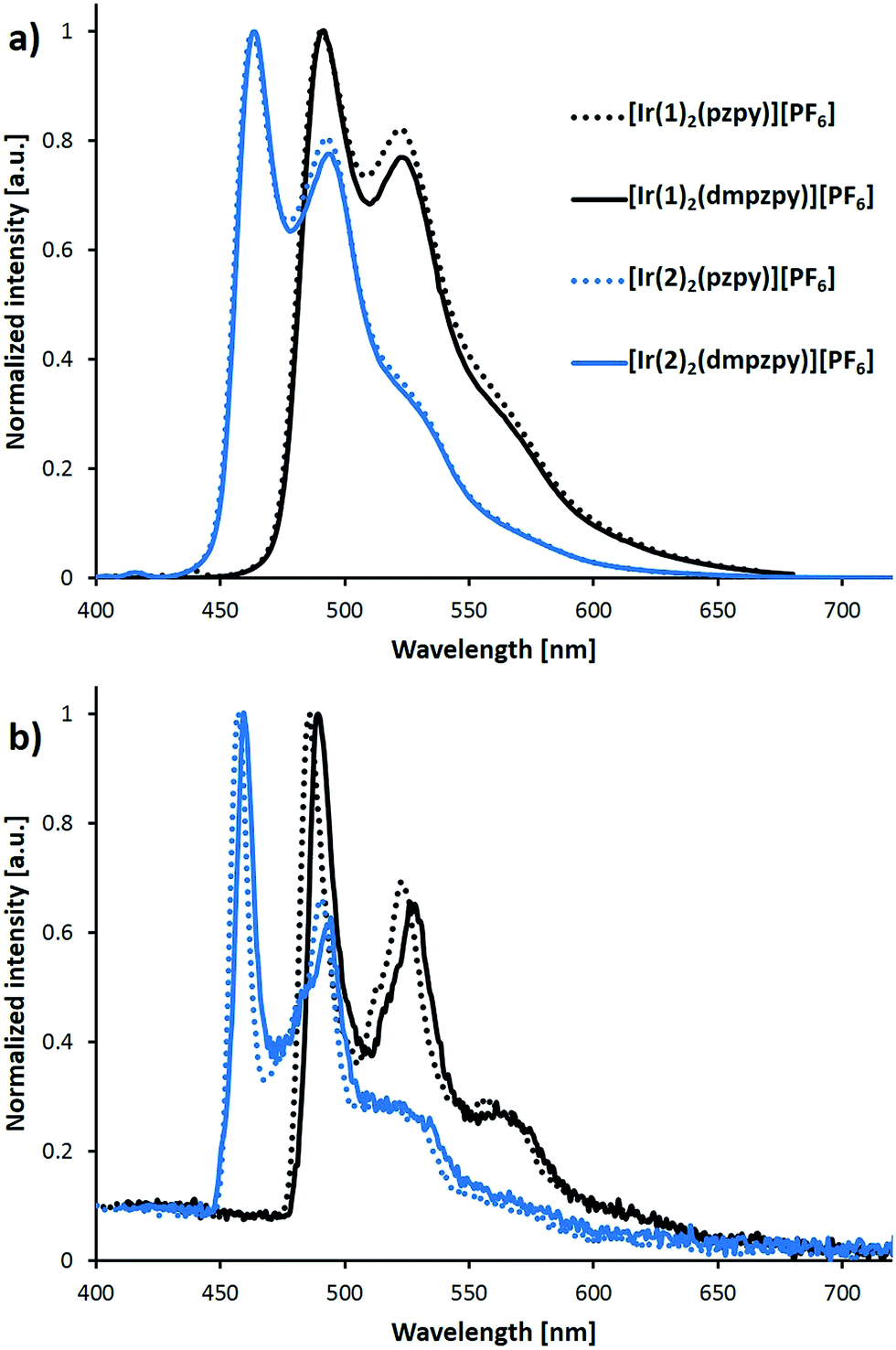 | ||
| Fig. 5 Photoluminescence spectra of complexes [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) recorded in MeCN solution (1 × 10−5 M) at room temperature (a) and at 77 K (b). Excitation wavelengths: 388 nm for [Ir(1)2(pzpy)][PF6], 350 nm for [Ir(1)2(dmpzpy)][PF6] and 370 nm for [Ir(2)2(N^N)][PF6]. At 77 K the excitation wavelength was fixed at 320 nm. Excitation wavelengths are different from those in Table 1 to avoid the first harmonic of the excitation in the spectrum; apart from this, emission spectra are independent of the excitation wavelengths used. | ||
| MeCN solution | Powder samples | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| At 298 K | At 77 K | At 298 K | |||||||
| Complex | λ maxem a [nm] | τ b [μs] | PLQYa [%] | k r [105 s−1] | k nr [105 s−1] | λ maxem c [nm] | λ maxem b [nm] | τ b [μs] | PLQYa [%] |
| a λ exc = 261 nm for [Ir(1)2(N^N)][PF6], 259 nm for [Ir(2)2(N^N)][PF6]. b λ exc = 280 nm. c λ exc = 320 nm. | |||||||||
| [Ir(1)2(pzpy)][PF6] | 491, 523 | 4.47 | 77 | 1.7 | 0.52 | 488, 526, 563 | 524 | 0.815 (97%), 2.48 (3%) | 7.5 |
| [Ir(1)2(dmpzpy)][PF6] | 491, 523 | 5.12 | 66 | 1.3 | 0.66 | 489, 526, 564 | 519 | 0.398 (89%), 1.00 (11%) | 5.9 |
| [Ir(2)2(pzpy)][PF6] | 463, 493 | 2.30 | 72 | 3.1 | 1.2 | 459, 492, 521 | 498 | 0.346 (90%), 1.35 (10%) | 4.6 |
| [Ir(2)2(dmpzpy)][PF6] | 463, 493 | 2.27 | 53 | 2.3 | 2.1 | 459, 494, 523 | 491 | 0.510 (94%), 1.33 (6%) | 11 |
Only a negligible hypsochromic shift of 2 nm in λmaxem occurs when the bpy ligand in [Ir(1)2(bpy)][PF6] (493 nm)31 is replaced with the electron-rich pzpy or dmpzpy. This suggests that the emissive state of [Ir(C^N)2(N^N)]+ complexes with cyclometallating ligand H1 is relatively independent of the N^N ligand and has a high 3LC character. In contrast, a blue-shift of >100 nm is observed on going from [Ir(ppy)2(bpy)][PF6] (585 nm)56 to [Ir(ppy)2(pzpy)][PF6] (475 nm).8 However, by changing the position of the sulfone group on the cyclometallating ligand, the emission maximum is blue-shifted by ≈30 nm from the green (complexes [Ir(1)2(N^N)][PF6]) to the blue region (complexes [Ir(2)2(N^N)][PF6]).
Photoluminescence quantum yields (PLQY, Table 1) of the complexes in de-aerated MeCN solution are in the range 53–77% and are comparable to the PLQY of [Ir(1)2(bpy)][PF6] (74%).31 [Ir(ppy)2(pzpy)][PF6], however, has a lower quantum yield of 23%.8 Shavaleev et al. have reported a series of iridium complexes with alkylated pyrazolylpyridine ligands.57 Strong phosphorescence quenching in solution was observed when the ancillary ligands contained a methyl group in the 5-position of the coordinated pyrazole ring; this may induce steric hindrance upon coordination to iridium. In the latter work, incorporation of the 5-Me substituent leads to a three-fold increase in the non-radiative decay rate (knr = (1 − PLQY)/τ), and the authors relate this to reversible dissociation of the sterically hindered Ir–N bond. This effect is only seen in solution and a visible emission from powdered samples of all their complexes was observed. In our series of complexes, however, no such large drop in PLQY is observed. Quantum yields of [Ir(C^N)2(dmpzpy)][PF6] with a methyl group in the 5-position of the pyrazole ring are only slightly lower than those of [Ir(C^N)2(pzpy)][PF6]. Calculated non-radiative decay rates knr of [Ir(C^N)2(dmpzpy)][PF6] are higher than for their non-substituted analogues, but the difference is less significant than in the previously reported examples.57 When solutions of [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) are not de-aerated, quantum yields decrease significantly to between 2.9 and 5.1%, consistent with oxygen quenching.
Excited-state lifetimes (τ) in de-aerated MeCN solution are of the order of a few microseconds for all the complexes. [Ir(2)2(N^N)][PF6] and [Ir(1)2(bpy)][PF6]31 have lifetimes of 2.3 μs, slightly longer than τ of [Ir(ppy)2(pzpy)][PF6] (1.56 μs).8 The complexes [Ir(1)2(N^N)][PF6] exhibit longer excited state lifetimes of around 5 μs. For [Ir(1)2(N^N)][PF6] and [Ir(2)2(dmpzpy)][PF6], the calculated radiative decay rate constants (kr = PLQY/τ) are between 1.3 × 105 and 2.3 × 105 s−1; these values compare to a kr value of 1.5 × 105 s−1 for [Ir(ppy)2(pzpy)][PF6].8 Only [Ir(2)2(pzpy)][PF6] has a higher kr of 3.1 × 105 s−1, comparable to [Ir(1)2(bpy)][PF6], with a value of 3.2 × 105 s−1.31 A higher kr indicates a lower 3LC character of the emissive state.1,8
Excitation of powder samples of [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) results in the emission spectra shown in Fig. 6, and PL data are summarized in Table 1. With respect to the solution spectra (Fig. 5), the solid-state emission maxima are red-shifted by about 30 nm and vibrational structure is less pronounced, suggesting strong intermolecular interactions in the solid state.8 Such a red-shift of the emission maximum on going from the solution to the solid seems to be common for blue or blue-green emitting iridium complexes. The extent of the red-shift can, however, vary between ≤15 nm8,28,58,59 and ≥33 nm.28,60,61
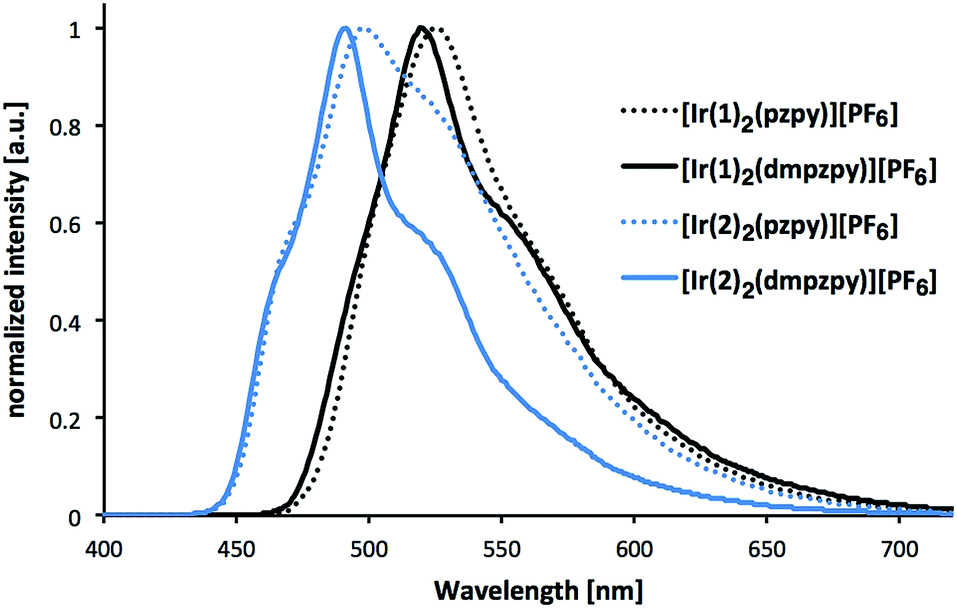 | ||
| Fig. 6 Photoluminescence spectra of complexes [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) in the solid state (powder). Excitation wavelength: 280 nm. | ||
In our previously reported sulfone-substituted [Ir(C^N)2(bpy)][PF6] complexes, red-shifts in the emission maxima of ≈40 nm were observed on going from solution to solid. In contrast, the yellow emitting fluorine- and thioether-containing [Ir(C^N)2(bpy)][PF6] in this series showed blue-shifts of 10–19 nm from solution to powder.31 Excited-state lifetimes (determined using second-order fits) in the solid state are much lower than in MeCN solution. In addition to the dramatically decreased PLQYs of powdered samples (≤11%), this suggests strong quenching of the excited state in powder samples.
Electrochemical properties
Cyclic voltammetric data for [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) are given in Table 2. All complexes show reversible oxidation processes. However, the first reduction is only reversible for [Ir(1)2(N^N)][PF6]. The HOMO is located on the cyclometallating ligand as well as the iridium(III) centre.1 Therefore, the similarity between Eox1/2 for [Ir(1)2(N^N)][PF6] and [Ir(1)2(bpy)][PF6] (+1.19, +1.20 and +1.18 V, respectively, with respect to Fc/Fc+) and for [Ir(2)2(N^N)][PF6] (+1.26 and +1.27 V) is as expected. The oxidation of each of [Ir(2)2(N^N)][PF6] (N^N = pzpy and dmpzpy) is shifted to slightly higher potential, consistent with stabilization of the HOMO. Compared to a value of −1.72 V for [Ir(1)2(bpy)][PF6],31 the first reduction occurs at lower potential (between −2.00 and −2.14 V). This is consistent with the replacement of bpy by the more electron-rich pyrazolylpyridine ligands leading to a destabilization of the LUMO. The larger electrochemical bandgap for complexes [Ir(2)2(N^N)][PF6] is consistent with the blue-shift of the emission compared to that of [Ir(1)2(N^N)][PF6].| Complex | E ox1/2 [V] | E red1/2 [V] | ΔE1/2a [V] |
|---|---|---|---|
| a ΔE1/2 = Eox1/2 − Ered1/2 | |||
| [Ir(1)2(pzpy)][PF6] | +1.19 | −2.02, −2.26ir, −2.68ir | 3.21 |
| [Ir(1)2(dmpzpy)][PF6] | +1.20 | −2.00, −2.27ir, −2.42ir, −2.63ir | 3.20 |
| [Ir(2)2(pzpy)][PF6] | +1.26 | −2.11ir, −2.37ir, −2.62ir | 3.37 |
| [Ir(2)2(dmpzpy)][PF6] | +1.27 | −2.14ir, −2.37qr, −2.68ir | 3.41 |
DFT/TD-DFT calculations
To gain a deeper insight into the electrochemical and photophysical properties of complexes [Ir(C^N)2(N^N)]+ (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy), a combined DFT/TD-DFT theoretical investigation was undertaken at the B3LYP/(6-31G** + LANL2DZ) level in the presence of solvent (acetonitrile). A similar study was recently performed for [Ir(ppy)2(pzpy)]+,62 which is used here as a reference for comparison purposes.Calculations predict a near-octahedral structure for the complexes in their ground electronic state (S0). The ancillary ligand is mostly planar in the pzpy complexes, the inter-annular N–N–C–N dihedral angle having a value of 3.4°. The presence of the methyl group in 5-position of the coordinated dmpzpy ligand introduces some steric hindrance and the N–N–C–N angle increases to 11.5°. This loss of planarity does not significantly affect the Ir–Npy and Ir–Npz coordination distances with the ancillary ligand, that change from 2.23 and 2.19 Å in the complexes with N^N = pzpy to 2.21 and 2.21 Å in those with N^N = dmpzpy, respectively.
Fig. 7 compares the energy and electron density contours calculated for the highest occupied (HOMO) and lowest-unoccupied molecular orbitals (LUMO and LUMO+1) of [Ir(ppy)2(pzpy)]+ with those obtained for [Ir(C^N)2(N^N)]+ (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy). The introduction of the sulfone groups leads to a stabilization of the HOMO, which is located, as expected, on the Ir atom and the phenyl rings of the cyclometallating ligands. The stabilization is slightly larger (0.08 eV) for the complexes with C^N = [2]− because of the higher participation of the carbon in 4-position to which the sulfone group is attached. The attachment of methyl groups to the N^N ligand has no relevant effect on the energy of the HOMO because this ligand does not contribute to the orbital. These trends explain the higher oxidation potentials measured for [Ir(2)2(N^N)]+ (+1.27 V) compared with [Ir(1)2(N^N)]+ (+1.20 V) and the negligible changes observed when substituting the pzpy ligand by dmpzpy (Table 2). The oxidation potential reported for [Ir(ppy)2(pzpy)]+ (+0.88 V)8 is significantly smaller in accord with the higher energy of its HOMO.
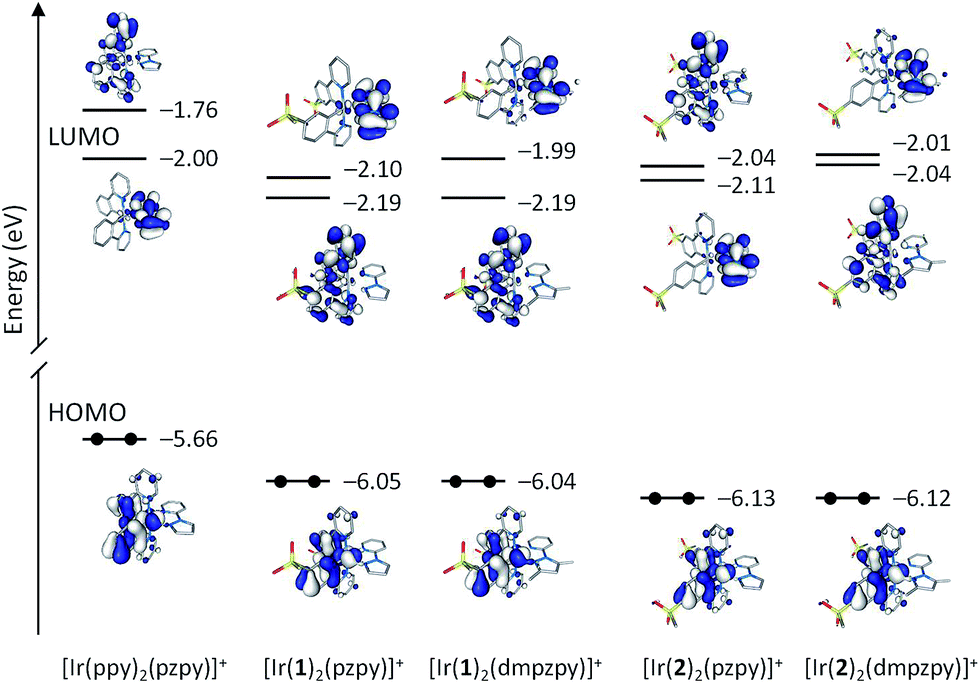 | ||
| Fig. 7 Schematic representation showing the isovalue contours (±0.03 a.u.) and energies calculated for the frontier molecular orbitals of [Ir(ppy)2(pzpy)]+ and complexes [Ir(C^N)2(N^N)]+ (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy). | ||
The LUMO of [Ir(ppy)2(pzpy)]+ is concentrated on the ancillary N^N ligand with a small contribution of the metal (Fig. 7). As for the HOMO, the introduction of the sulfone groups especially stabilizes the orbitals localized on the C^N ligands. As a result, the LUMO + 1 of [Ir(ppy)2(pzpy)]+ (−1.76 eV), which is centred on the C^N ligands, is lowered in energy by 0.43 eV for the [Ir(1)2(N^N)]+ complexes (−2.19 eV) and passes below the orbital centred on the N^N ligand and becomes the LUMO. For the [Ir(2)2(N^N)]+ complexes, the stabilization of the LUMO+1 of [Ir(ppy)2(pzpy)]+ is smaller (0.28 eV) due to the negligible participation of the carbon atom to which the sulfone group is attached, and the LUMO continues to be centred on the N^N ligand for [Ir(2)2(pzpy)]+ (Fig. 7). For [Ir(2)2(dmpzpy)]+, the addition of the methyl groups destabilizes the orbital centred on the N^N ligand by 0.10 eV and the LUMO again corresponds to the orbital centred on the C^N ligands. The energy of the LUMO therefore increases along the series [Ir(1)2(pzpy)]+: –2.19 eV = [Ir(1)2(dmpzpy)]+: −2.19 eV < [Ir(2)2(pzpy)]+: −2.11 eV < [Ir(2)2(dmpzpy)]+: −2.04 eV < [Ir(ppy)2(pzpy)]+: −2.00 eV, fully justifying the values recorded for the first reduction potential (−2.02 V ≈ −2.00 V > −2.11 V > −2.14 V > −2.19 V,8 respectively).
The combination of these effects leads to HOMO–LUMO energy gaps that increase along the series [Ir(ppy)2(pzpy)]+: 3.66 eV < [Ir(1)2(pzpy)]+: 3.86 eV ≈ [Ir(1)2(dmpzpy)]+: 3.85 eV < [Ir(2)2(pzpy)]+: 4.02 eV < [Ir(2)2(dmpzpy)]+: 4.08 eV. This trend correctly reproduces the evolution of the electrochemical gap (3.07 V8 < 3.21 V ≈ 3.20 V < 3.37 V < 3.41 V, respectively, Table 2) and the blue-shifted emission of complexes [Ir(2)2(N^N)]+ with respect to [Ir(1)2(N^N)]+ (Fig. 5). Indeed, if a direct correlation is established between the HOMO–LUMO gap and the emission energy, all the complexes should be expected to be bluer than [Ir(ppy)2(pzpy)]+. This is not however the case since the emission of [Ir(1)2(pzpy)]+ and [Ir(1)2(dmpzpy)]+ (λmaxem = 491, 523 nm, Table 1) is red-shifted compared to [Ir(ppy)2(pzpy)]+ (λmaxem = 475, 503 nm),8 whereas that of [Ir(2)2(pzpy)]+ and [Ir(2)2(dmpzpy)]+ (λmaxem = 463, 493 nm) is blue-shifted.
:[Bmim][PF6] 4:1 molar ratio [Bmim][PF6] = 1-butyl-3-methylimidazolium hexafluoridophosphate)
| Complex cation | t on a [s] | Lummaxb [cd m−2] | t 1/2 c [min] | PCEd [lm W−1] | EQEe [%] | λ maxEL [nm] |
|---|---|---|---|---|---|---|
| a Time to reach the maximum luminance. b Maximum luminance reached. c Time to reach one-half of the maximum luminance. d Power conversion efficiency (PCE). e External quantum efficiency (EQE). | ||||||
| [Ir(1)2(pzpy)]+ | 35 | 141 | 3.7 | 0.5 | 0.4 | 492, 530, 568 |
| [Ir(1)2(dmpzpy)] + | 78 | 125 | 6.2 | 0.6 | 0.4 | 492, 533, 568 |
| [Ir(2)2(pzpy)]+ | 42 | 49 | 3.5 | 0.2 | 0.2 | 466, 496, 547 |
| [Ir(2)2(dmpzpy)]+ | 30 | 129 | 2.4 | 0.5 | 0.4 | 466, 496, 547 |
To disentangle this apparent contradiction between theory and experiment and to obtain more information about the emitting state, the geometry of the lowest-energy triplet excited state T1 was optimized using the spin-unrestricted UB3LYP approach. Fig. 8a summarizes the adiabatic energy difference between S0 and T1 (ΔE) and the emission energy (Eem) estimated as the vertical energy difference between T1 and S0 at the optimized minimum-energy geometry of T1. Fig. 8b shows the unpaired-electron spin density distributions calculated for the lowest-energy triplet of complexes [Ir(1)2(pzpy)]+ and [Ir(2)2(pzpy)]+. Almost identical spin-density plots are obtained for complexes with N^N = dmpzpy and for [Ir(bpy)2(pzpy)]+. Therefore, independently of the nature of the LUMO, the spin-density distribution calculated for T1 is mainly centred on one of the cyclometallating ligands (∼1.65 unpaired electrons) with some contribution from the iridium atom (∼0.29e). This indicates that the lowest-energy triplet state has a predominant LC nature with some MLCT character in accord with the structured shape of the emission band observed experimentally.
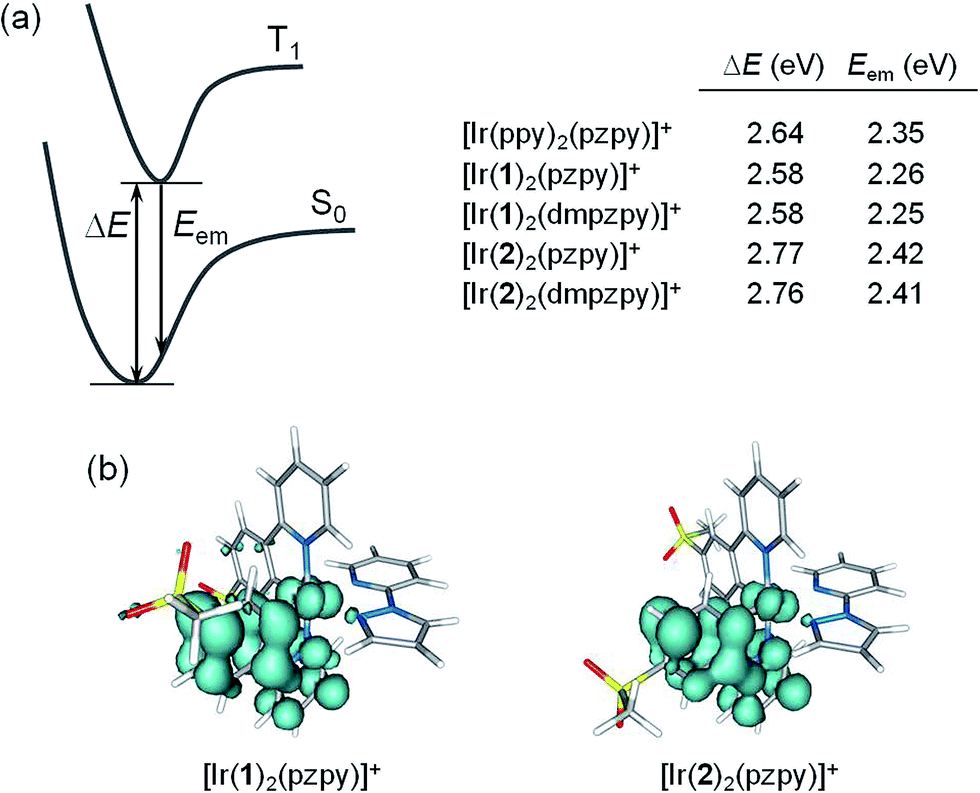 | ||
| Fig. 8 (a) Schematic diagram showing the adiabatic energy difference (ΔE) between S0 and T1 and the emission energy (Eem) from T1 computed for [Ir(bpy)2(pzpy)]+ and for complexes [Ir(C^N)2(N^N)]+ (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy). (b) Unpaired-electron spin density contours (0.002 au) calculated for the fully relaxed T1 state of [Ir(1)2(pzpy)]+ and [Ir(2)2(pzpy)]+. | ||
In agreement with experiment (Table 1), calculations find almost identical emission energies for complexes with C^N = [1]− (Eem ∼ 2.25 eV) and C^N = [2]− (Eem ∼ 2.41 eV). The emission energy is not affected by the addition of methyl groups to the ancillary ligand because the emission is centred on the C^N ligands (Fig. 8). Compared with [Ir(ppy)2(pzpy)]+ (Eem = 2.35 eV), the calculated emission energies correctly reproduce the red-shifted and blue-shifted emission observed experimentally for complexes [Ir(1)2(N^N)]+ and [Ir(2)2(N^N)]+, respectively.
Time dependent DFT (TD-DFT) calculations confirm the 3LC nature of the lowest-lying triplet (Table S1†). The first triplet state results from the excitation from the HOMO to the first virtual orbital located on the C^N ligands, i.e. the LUMO for [Ir(1)2(pzpy)]+, [Ir(1)2(dmpzpy)]+, and [Ir(2)2(dmpzpy)]+, and the LUMO + 1 for [Ir(ppy)2(pzpy)]+ and [Ir(2)2(pzpy)]+. A second 3LC triplet is found very close in energy (∼0.05 eV) due to the excitation from the HOMO to the second virtual MO centred on the C^N ligands. The first 3MLCT state is predicted to be higher than the 3LC state by an energy ranging from 0.2 eV in [Ir(ppy)2(pzpy)]+ to 0.6 eV in [Ir(1)2(dmpzpy)]+.
Electroluminescence
Complexes [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) were tested in LEC devices, which were operated using a pulsed current driving mode with a frequency of 1 kHz and a duty cycle of 50%.63,64 The LEC performance parameters measured using an average current density of 100 A m−2 are given in Table 3. Under these conditions, all the devices showed a typical LEC behaviour (Fig. S1–S4†). The voltage needed to maintain the established current density starts at a high value due to the initially high injection barriers and rapidly drops as a result of ion migration towards the electrodes forming an electric double-layer that reduces the injection barriers. The time needed to reach the maximum luminance (ton) is relatively short ranging from 30 to 78 seconds. The relatively fast response matches with the rather poor stability of the devices with lifetimes (t1/2) of a few minutes (Table 3). The results are comparable to those obtained for other blue-green LECs reported in the literature,12,16,22 for which a limited lifetime was observed especially when operated using a pulsed driving mode. In these cases, a relatively fast ton was also measured what is consistent with the values reported here.The maximum luminance achieved under pulsed current density of 100 A m−2 was 141 cd m−2 for the LEC prepared using [Ir(1)2(pzpy)][PF6]. The device performance is therefore not very high due to the moderate efficiencies. The trends obtained for the power conversion efficiency (PCE) and the external quantum efficiency (EQE) follow the photoluminescence quantum yields measured for thin films of the complexes with the same composition of the active layer in the device (Fig. S5†). Complex [Ir(2)2(pzpy)][PF6] with the lowest efficiency values (Table 3) show the smallest PLQY (6.9%), and the other three complexes with comparable higher efficiencies have PLQY values ranging from 13.0 to 16.9%.
Fig. 9 shows the electroluminescent (EL) spectra recorded for the LECs prepared. All the LECs exhibit structured electroluminescent emission bands coherent with the LC character of the emissive state. The position of the peaks forming the EL band are similar to the PL spectra depicted in Fig. 5 (compare Tables 1 and 3), but their relative intensity drastically changes. For complexes [Ir(2)2(N^N)][PF6], the EL spectra show bluer weak emission peaks at 466 and 496 nm and the maximum emission is at 546 nm (Fig. 9). For complexes [Ir(1)2(N^N)][PF6], an intense peak is observed at 492 nm but the maximum emission appears around 530 nm. The relative intensity of the EL peaks determines that the LEC devices exhibit a green colour for all the four complexes.
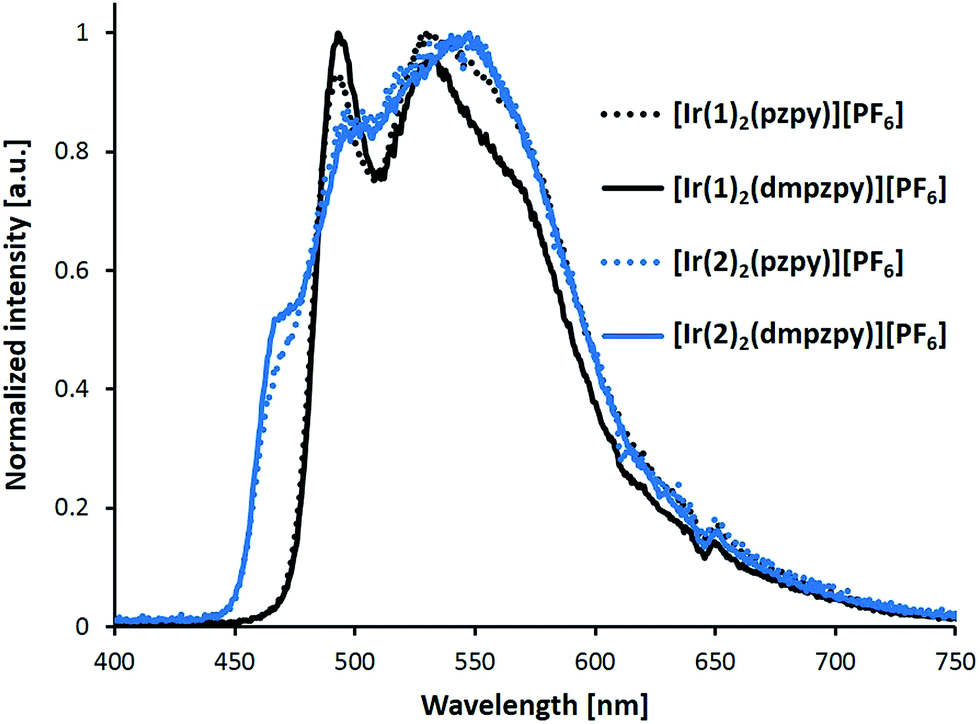 | ||
| Fig. 9 Electroluminescence spectra of LEC devices containing complexes [Ir(C^N)2(N^N)][PF6] (C^N = [1]− and [2]−, N^N = pzpy and dmpzpy) driven using a block-wave pulsed current of 100 A m−2 at a frequency of 1 kHz and duty cycles of 50%. | ||
Conclusions
A series of [Ir(C^N)2(N^N)]+ complexes with 2-phenylpyridine-based cyclometallating and pyrazolylpyridine ancillary ligands has been synthesized. Two different cyclometallating ligands, H1 and H2, with a sulfonyl substituent in either the 3- or 4-position of the phenyl ring of the ppy domain were combined with 2-(1H-pyrazol-1-yl)pyridine (pzpy) and 2-(3,5-dimethyl-1H-pyrazol-1-yl)pyridine (dmpzpy) N^N ligands. These ligand combinations were designed to blue-shift the emission of the [Ir(C^N)2(N^N)]+ complex. In practice, green or blue emissions were observed, the colour depending on the sulfone substituent position in the cyclometallating ligand.The complexes were characterized by spectroscopic and mass spectrometric methods, and the single crystal structure of [Ir(2)2(dmpzpy)][PF6]·MeCN has been determined. Excitation of the complexes in MeCN solution leads to structured emission spectra with maxima in the green region (491, 523 nm) for complexes with 2-(4-methylsulfonylphenyl)pyridine ([Ir(1)2(N^N)][PF6]) and in the blue region (463, 493 nm) for complexes with 2-(3-methylsulfonylphenyl)pyridine ([Ir(2)2(N^N)][PF6]). The marked vibrational structure and the absence of a rigidochromic shift in the low temperature spectra indicate significant 3LC contributions to the emissive state. Theoretical calculations show that the sulfone substituents strongly stabilize the orbitals located over the cyclometallating ligands and confirm the 3LC nature of the lowest-lying triplet state. PLQY values in de-aerated MeCN solution are quite high for all four complexes (between 53 and 77%), whereas the PLQY values of powdered samples are considerably lower than in solution (≤11%), suggesting strong quenching effects due to intermolecular interactions. Unfortunately, all four complexes performed poorly when tested in LEC configuration.
Methyl groups on the pyrazolylpyridine ligand did not have a significant effect on the properties of the complexes since both the emission and electrochemical properties are mainly determined by the cyclometallating ligands. On the other hand, changing the substitution position of the sulfone group on the cyclometallating ligand produced a shift of ≈30 nm in the emission maximum. This shift is reproduced in the theoretical study and is due to the different effect that sulfone substitution has on the frontier molecular orbitals depending on the substitution position. We are currently investigating the effect of substituental isomerism on the cyclometallating ligand in relation to colour tuning in more detail.
Acknowledgements
We thank the Swiss National Science Foundation (Grant number 200020_144500), the European Research Council (Advanced Grant 267816 LiLo) and the University of Basel for financial support. This work has also been supported by the European Community's Seventh Framework Programme (TREASORES Grant 314068 and LUMINET Grant 316906), the Spanish Ministry of Economy and Competitiveness (MINECO) (MAT2011-24594 and CTQ2012-31914), European Feder funds (CTQ2012-31914) and the Generalitat Valenciana (Prometeo/2012/053). Lucas A. Hauser is thanked for help with synthesis and characterization of complexes [Ir(1)2(N^N)][PF6]. Dr Alessandro Prescimone is thanked for determining the preliminary crystal structure of complex [Ir(1)2(dmpzpy)][PF6].Notes and references
- R. D. Costa, E. Ortí, H. J. Bolink, F. Monti, G. Accorsi and N. Armaroli, Angew. Chem., Int. Ed., 2012, 51, 8178 CrossRef CAS PubMed and references threrein.
- T. Hu, L. He, L. Duan and Y. Qiu, J. Mater. Chem., 2012, 22, 4206 RSC.
- Q. Pei, G. Yu, C. Zhang, Y. Yang and A. J. Heeger, Science, 1995, 269, 1086 CrossRef CAS PubMed.
- J. C. deMello, N. Tessler, S. C. Graham and R. H. Friend, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 57, 12951 CrossRef CAS.
- J. D. Slinker, J. A. DeFranco, M. J. Jaquith, W. R. Silveira, Y. Zhong, J. M. Moran-Mirabal, H. G. Graighead, H. D. Abruña, J. A. Marohn and G. G. Malliaras, Nat. Mater., 2007, 6, 894 CrossRef CAS PubMed.
- Md. K. Nazeeruddin, R. T. Wegh, Z. Zhou, C. Klein, Q. Wang, F. De Angelis, S. Fantacci and M. Grätzel, Inorg. Chem., 2006, 45, 9245 CrossRef CAS PubMed.
- A. B. Tamayo, S. Garon, T. Sajoto, P. I. Djurovich, I. M. Tsyba, R. Bau and M. E. Thompson, Inorg. Chem., 2005, 44, 8723 CrossRef CAS PubMed.
- L. He, L. Duan, J. Qiao, R. Wang, P. Wei, L. Wang and Y. Qiu, Adv. Funct. Mater., 2008, 18, 2123 CrossRef CAS.
- L. He, L. Duan, J. Qiao, D. Zhang, L. Wang and Y. Qiu, Chem. Commun., 2011, 47, 6467 RSC.
- L. He, J. Qiao, L. Duan, G. Dong, D. Zhang, L. Wang and Y. Qiu, Adv. Funct. Mater., 2009, 19, 2950 CrossRef CAS.
- B. Chen, Y. Li, W. Yang, W. Luo and H. Wu, Org. Electron., 2011, 12, 766 CrossRef CAS.
- M. Mydlak, C. Bizzarri, D. Hartmann, W. Sarfert, G. Schmid and L. De Cola, Adv. Funct. Mater., 2010, 20, 1812 CrossRef CAS.
- C.-H. Yang, J. Beltran, V. Lemaur, J. Cornil, D. Hartmann, W. Sarfert, R. Frohlich, C. Bizzarri and L. De Cola, Inorg. Chem., 2010, 49, 9891 CrossRef CAS PubMed.
- L. He, L. Duan, J. Qiao, G. Dong, L. Wang and Y. Qiu, Chem. Mater., 2010, 22, 3535 CrossRef CAS.
- T. Hu, L. Duan, J. Qiao, L. He, D. Zhang, R. Wang, L. Wang and Y. Qiu, Org. Electron., 2012, 13, 1948 CrossRef CAS.
- S. B. Meier, W. Sarfert, J. M. Junquera-Hernández, M. Delgado, D. Tordera, E. Ortí, H. J. Bolink, F. Kessler, R. Scopelliti, M. Grätzel, M. K. Nazeeruddin and E. Baranoff, J. Mater. Chem. C, 2013, 1, 58 RSC.
- H.-C. Su, H.-F. Chen, P.-H. Chen, S.-W. Lin, C.-T. Liao and K.-T. Wong, J. Mater. Chem., 2012, 22, 22998 RSC.
- E. Baranoff, H. J. Bolink, E. C. Constable, M. Delgado, D. Häussinger, C. E. Housecroft, M. K. Nazeeruddin, M. Neuburger, E. Ortí, G. E. Schneider, D. Tordera, R. M. Walliser and J. A. Zampese, Dalton Trans., 2013, 1073 RSC.
- L. He, L. Dian, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. Qiu, Synth. Met., 2013, 166, 52 CrossRef CAS.
- J. M. Fernández-Hernández, S. Ladouceur, Y. Shen, A. Iordache, X. Wang, L. Donato, S. Gallagher-Duval, M. de Anda Villa, J. D. Slinker, L. De Cola and E. Zysman-Colman, J. Mater. Chem. C, 2013, 1, 7440 RSC.
- C. D. Sunesh, K. Shanmugasundaram, M. S. Subeesh, R. K. Chitumalla, J. Jang and Y. Choe, ACS Appl. Mater. Interfaces, 2015, 7, 7741 CAS.
- F. Monti, A. Baschieri, I. Gualandi, J. J. Serrano-Pérez, J. M. Junquera-Hernández, D. Tonelli, A. Mazzanti, S. Muzzioli, S. Stagni, C. Roldán-Carmona, A. Pertegás, H. J. Bolink, E. Ortí, L. Sambri and N. Armaroli, Inorg. Chem., 2014, 53, 7709 CrossRef CAS PubMed.
- D. Tordera, J. J. Serrano-Pérez, A. Pertegás, E. Ortí, H. J. Bolink, E. Baranoff, Md. K. Nazeeruddin and J. Frey, Chem. Mater., 2013, 25, 3391 CrossRef CAS.
- See for example: C.-H. Lin, Y.-Y. Chang, J.-Y. Hung, C.-Y. Lin, Y. Chi, M.-W. Chung, C.-L. Lin, P.-T. Chou, G.-H. Lee, C.-H. Chang and W.-C. Lin, Angew. Chem., Int. Ed., 2011, 50, 3182 CrossRef CAS PubMed; V. Sivasubramaniam, F. Brodkorb, S. Hanning, H. P. Loebl, V. van Elsbergen, H. Boerner, U. Scherf and M. Kreyenschmidt, J. Fluorine Chem., 2009, 130, 640 CrossRef.
- H. J. Bolink, L. Cappelli, S. Cheylan, E. Coronado, R. D. Costa, N. Lardiés, Md. K. Nazeeruddin and E. Ortí, J. Mater. Chem., 2007, 17, 5032 RSC.
- H. Oh, K.-M. Park, H. Hwang, S. Oh, J. H. Lee, J.-S. Lu, S. Wang and Y. Kang, Organometallics, 2013, 32, 6427 CrossRef CAS.
- J. Frey, B. F. E. Curchod, R. Scopelliti, I. Tavernelli, U. Rothlisberger, K. N. Mohammad and E. D. Baranoff, Dalton Trans., 2014, 5667 RSC.
- S. Evariste, M. Sandroni, T. W. Rees, C. Roldán-Carmona, L. Gil-Escrig, H. J. Bolink, E. Baranoff and E. Zysman-Colman, J. Mater. Chem. C, 2014, 2, 5793 RSC.
- See for example: M. Tavasli, S. Bettington, I. F. Perepichka, A. S. Batsanov, M. R. Bryce, C. Rothe and A. P. Monkman, Eur. J. Inorg. Chem., 2007, 4808 CrossRef CAS; G. Zhou, C.-L. Ho, W.-Y. Wong, Q. Wang, D. Ma, L. Wang, Z. Lin, T. B. Marder and A. Beeby, Adv. Funct. Mater., 2008, 18, 499 CrossRef; R. Ragni, E. Orselli, G. S. Kottas, O. H. Omar, F. Babudri, A. Pedone, F. Naso, G. M. Farinola and L. De Cola, Chem.–Eur. J., 2009, 15, 136 CrossRef PubMed; Y. Hisamatsu and S. Aoki, Eur. J. Inorg. Chem., 2011, 5360 CrossRef; C. Fan, Y. Li, C. Yang, H. Wu, J. Qin and Y. Cao, Chem. Mater., 2012, 24, 4581 CrossRef.
- D. Tordera, A. M. Bünzli, A. Pertegás, J. M. Junquera-Hernández, E. C. Constable, J. A. Zampese, C. E. Housecroft, E. Ortí and H. J. Bolink, Chem.–Eur. J., 2013, 19, 8597 CrossRef CAS PubMed.
- E. C. Constable, C. D. Ertl, C. E. Housecroft and J. A. Zampese, Dalton Trans., 2014, 5343 RSC.
- Y. You and S. Y. Park, Dalton Trans., 2009, 1267 RSC.
- H. A. Bronstein, C. E. Finlayson, K. R. Kirov, R. H. Friend and C. K. Williams, Organometallics, 2008, 27, 2980 CrossRef CAS.
- T.-H. Kwon, H. S. Cho, M. K. Kim, J.-W. Kim, J.-J. Kim, K. H. Lee, S. J. Park, I.-S. Shin, H. Kim, D. M. Shin, Y. K. Chung and J.-I. Hong, Organometallics, 2005, 24, 1578 CrossRef CAS.
- Q.-L. Xu, C.-C. Wang, T.-Y. Li, M.-Y. Teng, S. Zhang, Y.-M. Jing, X. Yang, W.-N. Li, C. Lin, Y.-X. Zheng, J.-L. Zuo and X.-Z. You, Inorg. Chem., 2013, 52, 4916 CrossRef CAS PubMed.
- X. Qu, Y. Liu, G. Godefroid, Y. Si, X. Shang, X. Wu and Z. Wu, Eur. J. Inorg. Chem., 2013, 3370 CrossRef CAS.
- APEX2, version 2 User Manual, M86-E01078, Bruker Analytical X-ray Systems, Inc., Madison, WI, 2006 Search PubMed.
- P. W. Betteridge, J. P. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999 CrossRef CAS PubMed.
- J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027 CrossRef CAS.
- C. S. Cramer and D. G. Truhlar, in Solvent Effects and Chemical Reactivity, Kluwer, Dordrecht, 1996, pp. 1–80 Search PubMed.
- M. Petersilka, U. J. Gossmann and E. K. U. Gross, Phys. Rev. Lett., 1996, 76, 1212 CrossRef CAS PubMed.
- M. E. Casida, C. Jamorski, K. C. Casida and D. R. Salahub, J. Chem. Phys., 1998, 108, 4439 CrossRef CAS.
- C. Jamorski, M. E. Casida and D. R. Salahub, J. Chem. Phys., 1996, 104, 5134 CrossRef CAS.
- C. Liu, Q. Ni, P. Hu and J. Qiu, Org. Biomol. Chem., 2011, 9, 1054 CAS.
- H. Zhang, Q. Cai and D. Ma, J. Org. Chem., 2005, 70, 5164 CrossRef CAS PubMed.
- S. Sprouse, K. A. King, P. J. Spellane and R. J. Watts, J. Am. Chem. Soc., 1984, 106, 6647 CrossRef CAS.
- E. Baranoff, B. F. E. Curchod, J. Frey, R. Scopelliti, F. Kessler, I. Tavernelli, U. Rothlisberger, M. Grätzel and M. K. Nazeeruddin, Inorg. Chem., 2012, 51, 125 Search PubMed.
- F. A. M. Rudolph, A. L. Fuller, A. M. Z. Slawin, M. Bühl, R. A. Aitken and J. D. Woollins, J. Chem. Crystallogr., 2010, 40, 253 CrossRef CAS.
- C. Glidewell, W. T. A. Harrison, J. N. Low, J. G. Sime and J. L. Wardell, Acta Crystallogr., Sect. B: Struct. Sci., 2001, 57, 190 CAS.
- R. D. Costa, E. Ortí, D. Tordera, A. Pertegás, H. J. Bolink, S. Graber, C. E. Housecroft, L. Sachno, M. Neuburger and E. C. Constable, Adv. Energy Mater., 2011, 1, 282 CrossRef CAS.
- N. M. Shavaleev, R. Scopelliti, M. Grätzel and M. K. Nazeeruddin, Inorg. Chim. Acta, 2013, 404, 210 CrossRef CAS.
- F. Zhang, L. Duan, J. Qiao, G. Dong, L. Wang and Y. Qiu, Org. Electron., 2012, 13, 1277 CrossRef CAS.
- X. Qu, Y. Liu, Y. Si, X. Wu and Z. Wu, Dalton Trans., 2014, 1246 RSC.
- F. Zhang, D. Ma, L. Duan, J. Qiao, G. Dong, L. Wang and Y. Qiu, Inorg. Chem., 2014, 53, 6596 CrossRef CAS PubMed.
- C. D. Sunesh, G. Mathai and Y. Choe, Org. Electron., 2014, 15, 667 CrossRef CAS.
- P. Pla, J. M. Junquera-Hernández, H. J. Bolink and E. Ortí, Dalton Trans., 2015, 44, 8497 RSC.
- D. Tordera, A. M. Bünzli, A. Pertegás, J. M. Junquera-Hernández, E. C. Constable, J. A. Zampese, C. E. Housecroft, E. Ortí and H. J. Bolink, Chem.–Eur. J., 2013, 19, 8597 CrossRef CAS PubMed.
- D. Tordera, S. Meier, M. Lenes, R. D. Costa, E. Ortí, W. Sarfert and H. J. Bolink, Adv. Mater., 2012, 24, 897 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental details; syntheses of ligands, and IR spectroscopic data for complexes. Table S1: results of TD-DFT calculations. Fig. S1–S4: electroluminescent device data; Fig. S5: PL spectra of complexes in thin film. CCDC 1055780. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra07940c |
| This journal is © The Royal Society of Chemistry 2015 |