
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supramolecular host–guest complexation of Lash's calix[4]azulene with tetraalkylammonium halides and tetrafluoroborate salts: binding and DFT computational studies†
Shofiur
Rahman
a,
Ahmed
Zein
a,
Louise N.
Dawe
b,
Grigory
Shamov
c,
Pall
Thordarson
d and
Paris E.
Georghiou
*a
aDepartment of Chemistry, Memorial University of Newfoundland, St. John's, Newfoundland and Labrador, Canada A1B3X7. E-mail: parisg@mun.ca
bDepartment of Chemistry and Biochemistry, Wilfrid Laurier University, Waterloo, Ontario, Canada N2L 3C5
cWestgrid/ComputeCanada, University of Manitoba, Winnipeg, Manitoba, Canada R3T 2N2
dSchool of Chemistry and the ARC Centre for Excellence in Convergent Bio-Nano Science and Technology, University of New South Wales, Sydney 2052, Australia
First published on 16th June 2015
Abstract
Calix[4]azulene 1 is shown to be an effective molecular receptor for tetraalkylammonium halide and BF4− salts. The respective binding constants were determined using global-fit analyses of the spectral data from UV-vis absorption titration studies. A DFT study of the putative complexes formed with 1, and the X-ray structure of 1 itself is also reported.
Introduction
In 2002 Colby and Lash1 reported their remarkably facile synthesis of calix[4]azulene 1 by the Florisil-mediated reaction of azulene 2 (Fig. 1) with paraformaldehyde in dichloromethane. Since then, there have been no reports published on any of the potential supramolecular or molecular recognition properties of this intriguing compound.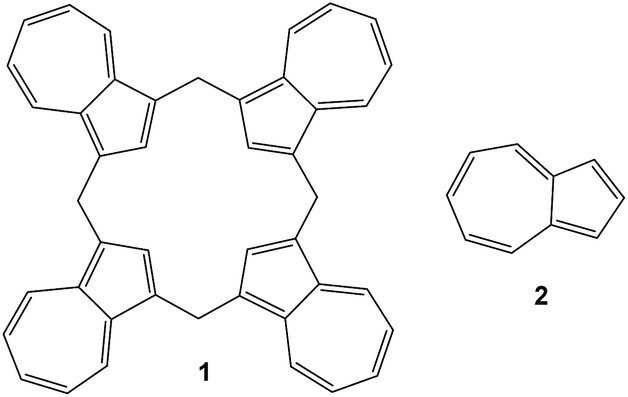 | ||
| Fig. 1 Structures of calix[4]azulene 1 and azulene 2. | ||
Due to the resemblance of 1 to calix[4]arenes2 the potential for molecular recognition studies with these compounds is an obvious one and due to our own on-going interest in the supramolecular complexation properties of such deep-cavity containing molecular receptors.3a–d We report here our findings with respect to a supramolecular host–guest complexation study of 1 with, in particular, tetraalkylammonium halides and tetrafluoroborate salts. A global analytical fitting4a,b was used to determine the binding or association constants from UV-vis titration experiments. During the course of our investigations, crystals of calix[4]azulene from benzene solution were obtained which permitted an X-ray structural determination.5 Concurrently, a DFT (Density Functional Theory)6 computational study was also conducted on both 1 and its complexes.
Results and discussion
In connection with our on-going studies of the supramolecular complexation properties of macrocyclic compounds, we were interested in evaluating those of 1. Our own studies have included the supramolecular binding, or complexation, of tetraalkyl-ammonium salts with various macrocyclic cavitands.3a–d Calix[4]azulene, on the other hand, is a hydrocarbon compound which among other significant differences, and when compared with other macrocyclic compounds which we have studied, has no heteroatoms in its structure. We were therefore interested in seeing whether this would affect the molecular receptor or binding properties of calix[4]azulene.The ambient temperature 1H NMR spectrum of 1 shows that the methylene bridge protons appear as a sharp singlet at δ = 4.74 ppm, indicating its conformational flexibility. However, in principle, 1 could potentially adopt cone, partial cone, 1,3- or 1,2-alternate conformations which are analogous to those that are typically associated with calix[4]arenes.2 The relatively low solubility of 1 in the usual NMR solvents however, prevented the determination of a coalescence temperature or energy using low-temperature VT-1H NMR spectroscopy.
Since 1 is a hydrocarbon molecule, we hypothesized that its macrocyclic cavity could serve as a site for molecular recognition with, in particular, aromatic hydrocarbon guest molecules, including e.g. naphthalene, hexamethylbenzene and C60 and C70. However using 1H NMR titration7 experiments no meaningful chemical shift changes for either the guest molecules or 1 were observed. Hence no evidence could be observed for any supramolecular complexation of these types of guests with 1.
Nevertheless, a mole ratio plot from a 1H-NMR titration experiment with tetramethylammonium chloride (TMACl) in CDCl3 as a guest, did reveal that a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 complex of TMACl with 1 formed. The low solubilities however, of both host and guest, precluded accurate determinations of the binding or association constant (Kassoc) in subsequent titration experiments. UV-vis spectroscopic titration experiments nevertheless proved more suitable for the studies with TMACl and the other tetraalkylammonium salts reported herein. It should be noted that although fluorescence spectroscopy is more sensitive than UV-vis spectroscopy, and could therefore be used with more dilute (∼10−5 to 10−3 M) solutions than those needed for either 1H-NMR or UV-vis titrations, fluorescence titration experiments could not be used with calix[4]azulene.8–10
:1 complex of TMACl with 1 formed. The low solubilities however, of both host and guest, precluded accurate determinations of the binding or association constant (Kassoc) in subsequent titration experiments. UV-vis spectroscopic titration experiments nevertheless proved more suitable for the studies with TMACl and the other tetraalkylammonium salts reported herein. It should be noted that although fluorescence spectroscopy is more sensitive than UV-vis spectroscopy, and could therefore be used with more dilute (∼10−5 to 10−3 M) solutions than those needed for either 1H-NMR or UV-vis titrations, fluorescence titration experiments could not be used with calix[4]azulene.8–10
X-ray crystallography of 1
During the course of our investigations, green crystals of 1 were obtained from benzene solution which were suitable for a single-crystal X-ray determination. The molecule crystallized in C2/c, with half of one molecule contained in the asymmetric unit (see Fig. 2a for the symmetry-expanded full molecule). Each plane made by C1–C10 and C12–C21 was oriented at 86.27(6)° to the next plane, with an inversion centre located directly in the middle of the molecule, leading to an anti-arrangement (or Ci-symmetrical conformer) for calix[4]azulene. There was no associated lattice solvent present. However, examination of the packing revealed close associations between the C1–C10 planes of adjacent molecules (plane-to-plane separation of 3.4397(17) Å, off-set by 3.496(3) Å) leading to a staircase arrangement of the molecules parallel to the b-axis, as shown in the space-filling Fig. 2b.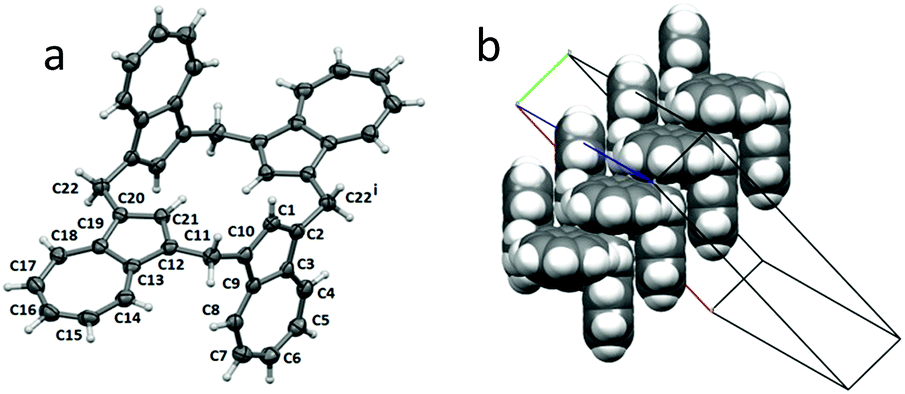 | ||
| Fig. 2 Calix[4]azulene (a) represented as 50% displacement ellipsoids. Symmetry operation (i) = 1/2 − x, 1.5 − y, 1 − z; and (b) by a space-filling representation showing the staircase arrangement of molecules parallel to the b-axis. | ||
UV-vis absorption studies
The UV-vis titration behaviour of 1 with tetramethylammonium halides (TMAX, X = Cl−, Br− and I−) could be followed despite the solubility limitations posed by both calixazulene and the TMAX salts. This was achieved using 10 cm pathlength cells in a thermostated dual-beam spectrophotometer. Addition of aliquots of the respective TMAX salts to solutions of 1 (4.8 × 10−5 M) resulted in a quenching of the spectra of 1 in the range of 525–700 nm. Although these quenching changes were small, they were sufficient to allow the resulting Kassoc values to be determined. Each of the individual full spectra that were obtained from duplicate titrations were subjected to a non-linear 1:1 global fit analysis.4b Average Kassoc ± average symptotic “fitting” uncertainty (ASFU) values of 7400 ± 0.44%, 3820 ± 1.3% and 2840 ± 1.2% M−1 respectively, for TMACl, TMABr and TMAI were obtained. The trend in Kassoc values between these halides is consistent with those seen in other instances.3a–d The global analysis approach provides a more accurate description of the equilibria involved since it avoids the manipulation of the actual experimental data to effect linear transformations of non-linear phenomena, and also that quality-of-fit parameters are also generated. The limitations of the classical double-reciprocal Benesi–Hildebrand treatment11 has been well-documented by others, including ourselves.4,12
To evaluate the potential effects of other larger tetraalkylammonium groups, the effects of higher homologues of TMAX, namely those of the corresponding tetraethyl and tetra-n-butylammonium salts were examined where possible. Solubility problems encountered with these higher tetraalkylammonium halide homologues, were averted by comparing TMABF4 with tetraethylammonium tetrafluoroborate (TEABF4), and tetra-n-butylammonium tetrafluoroborate (TBABF4) since these salts had higher solubilities in the 9:1 (v/v) CHCl3:CH3OH mixed solvent which was the solvent used for all of the UV-vis titrations. The resulting Kassoc values are shown in Table 1, entries 4–6. The trend observed, namely TMABF4 > TEABF4 > TBABF4 is consistent with the DFT calculations shown in Table 3.
| Entry | K assoc (M−1) | |
|---|---|---|
| Average ± ASFU | ||
| 1 | TMACl | 7400 ± 0.44% |
| 2 | TMABr | 3820 ± 1.3% |
| 3 | TMAI | 2840 ± 1.2% |
| 4 | TMABF4 | 5920 ± 1.2% |
| 5 | TEABF4 | 5060 ± 1.5% |
| 6 | TBABF4 | 3620 ± 1.3% |
DFT calculation details
The use of DFT has become increasingly commonplace in organic chemistry6,13 and among supramolecular chemists in particular. In this work we modeled complexation of the TMA halides and TMABF4 TEABF4, TBABF4 “guests” with the calixazulene host in the gas phase as well as in chloroform solution. All of the DFT calculations were conducted with the Gaussian-09 rev D.01 code.14For the work reported herein, Chai and Head-Gordon's ωB97xD15 functional, which combines the long-range functional ωB97x with the empirical dispersion correction specially parametrized against each other, was used. B3LYP results are provided for comparison purposes only. The popular B3LYP density functional16 which has often been employed by organic chemists has been shown by many authors, including those cited in ref. 17–20, to have numerous shortcomings. These shortcomings are related to the lack of dispersion interactions and the deficiencies of DFT with self-interaction errors and long-range behavior. In the last decade, in order to amend the poor performance of standard DFT, the following methods were proposed, and have become widely used: the empirical dispersion corrections by Grimme,17,21,22 and the long-range hybrid density functionals of Tsuneda and Hirao20 that, when combined, deliver much improved performance for thermodynamics and structure optimization. The effect is most pronounced for host-guest complexes such as were studied in our work where the interactions are dominated by non-bonded terms.22
The standard 6-31G(d) basis set23 was used for all the atoms. This basis set is small by modern standards but we chose to use it due to the relatively large size of our systems. It was shown that larger basis sets including diffuse functions cause basis set overcompleteness problems for condensed hydrocarbons. In agreement with Fry24 we found that the 6-31G(d) basis set was not reliable for the energies of complexes containing bromide anion; for iodide, non-relativistic all-electron calculations are not accurate. Therefore, for guests and complexes containing atoms other than first row elements (i.e. for TMACl, TMABr, TMAI) we used relativistic ECPs by Hay and Wadt (LANL) along with corresponding LANL2DZ basis set augmented with additional d-, p-polarizational functions.25,26a–c Cartesian Gaussian functions (6D, 7F) were used for the halides' LANL2DZ basis set, to make it consistent with the default setting for the 6-31G(d) basis set. For each of the individual components i.e. tetraalkylammonium salt, calix[4]azulene and the respective corresponding 1:1 supramolecular complexes, unconstrained geometry optimizations were first conducted in the gas phase. Then, geometries were optimized within the continuum solvation model (PCM)27a,b of the chloroform solvent, using default solvent parameters as provided with Gaussian-09 rev D.01. The results are summarized in Tables 2 and 3.
| B3LYP/6-31G(d) | kJ mol−1 | B3LYP/6-31G(d) | kJ mol−1 |
|---|---|---|---|
| Gas phase | CHCl 3 | ||
| C i | 0 | C i | 0 |
| C 2v Chair | +0.34 | C 2v-1,3-alternate | +0.73 |
| C 2v-1,3-alternate | +0.68 | C 2v chair | +0.33 |
| ωB97xD/6-31G(d) | kJ mol−1 | ωB97xD/6-31G(d) | kJ mol−1 |
|---|---|---|---|
| Gas phase | CHCl 3 | ||
| C s | 0 | C s | 0 |
| C 2v-1,3-alternate | +0.58 | C *s | 0 |
| C i | +10.1 | C i | +8.94 |
 | ||
| Fig. 3 Structures and symmetries of the most stable conformers of calix[4]azulene (all hydrogen atoms have been omitted for clarity) from the geometry-optimizations using (a) Ci-symmetry from the B3LYP/6-31G(d) geometry-optimization; and (b) Cs-symmetry from the ωB97xD/6-31G(d) geometry-optimization. | ||
Inclusion of chloroform solvation does not affect the relative energies of the conformers it can be seen that for B3LYP/6-31G(d) the energy difference between the three studied conformers is negligible, while ωB97xD/6-31G(d) has a marked preference for the Cs-symmetrical “flattened” 1,3-alternate conformer, which can be understood by favourable intra-molecular dispersion interaction between the parallel azulene rings in the isolated molecule. These interactions are absent in B3LYP but are included in the ωB97xD functional. The experimentally observed structure shown in Fig. 2a is probably a result of crystal packing forces. In Table 2, the most stable of the conformers for each of the density functionals were used to compute the “binding” energies of the complexes shown. In one case (shown in Table 2 by the entry marked as C*s) where the ωB97xD/6-31G(d) geometry-optimized C2v-1,3-alternate conformer from the gas phase was subjected to the ωB97xD/6-31G(d) geometry-optimization with the chloroform PCM the resulting conformer had the same Cs-symmetry.
:1 calix[4]azulene:TMA halide complexes their geometry-optimized conformers were found to exist with two potential energy surface minima: 1,3-alternate (shown in Fig. 4a and b) and partial cone (Fig. 4c). The B3LYP/6-31G(d) method initially found only the 1,3-alternate structures for all three halides; however, according to results from the ωB97xD functionals, this structure is more stable for TMACl and TMABr, while for TMAI, a partial cone conformer is preferred by 12.3 kJ mol−1. For the tetraalkylammonium tetrafluoroborate salts (TMABF4, TEABF4, TBABF4) geometry optimizations converged to the partial cone for both of the density functionals. Inclusion of the PCM chloroform solvation did not change the energy order of the isomers.
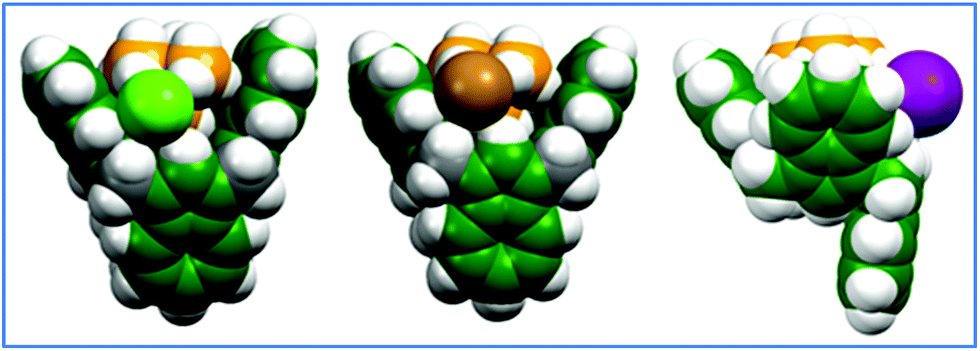 | ||
| Fig. 4 Geometry-optimized (ωB97xD/6-31G(d)) structures computed for: (a) left: TMACl ⊂ 1; (b) middle: TMABr ⊂ 1; (c) right: TMAI ⊂ 1. | ||
The “binding” or “interaction” (negative) energies (“BE”) generally decreased in magnitude (i.e. less energetically favoured) in going from the ωB97xD/6-31G(d) gas-phase to the CHCl3-corrected computed values, to the corresponding B3LYP/6-31G(d)-computed values, as summarized in Table 3. On the other hand, the binding constants observed for the tetramethyl-ammonium halides showed the trend: Cl− > Br− > I−. The BE of the complexes were calculated according to eqn (1) where Ecalixazulene is the geometry-optimized energy of calix[4]-azulene 1 and Etetraalkylammonium salt is that of the respective tetraalkylammonium salt. Ecomplex is the geometry-optimized energy of the complex formed from 1 with the respective tetraakylammonium salts.
| BE = Ecomplex − Σ(Ecalixazulene + Etetraalkylammonium salt) | (1) |
| ωB97xD/6-31G(d) | B3LYP/6-31G(d) | |||
|---|---|---|---|---|
| Gas phase (kJ mol−1) | CH3Cl (kJ mol−1) | Gas phase (kJ mol−1) | CH3Cl (kJ mol−1) | |
| a Note: the values in parentheses are the BEs of the TMAI complexes computed as the corresponding 1,3-alternate conformer of 1. | ||||
| TMACl | −113.9 | −77.3 | −33.2 | −13.1 |
| TMABr | −118.3 | −80.8 | −34.4 | −14.4 |
| TMAI | −133.5 (−121.2)a | −93.0 (−84.0)a | −35.4 | −13.9 |
| TMABF4 | −144.6 | −116.9 | −49.9 | −20.8 |
| TEABF4 | −129.0 | −111.9 | −29.7 | −8.3 |
| TBABF4 | −113.5 | −115.1 | −25.6 | −10.1 |
For the halides and BF4−, inclusion of the solvation effects does not change the trends but decreases the BEs uniformly; this is as it should be due to the relative stabilization of the isolated guest which is more polar. The B3LYP density functional predicts significantly weaker binding energies for missing dispersion interactions. Moreover, it does not reveal much difference in binding between the three TMA halide complexes. The more reliable ωB97xD functional predicts increases in the complexes' stability in the order of Cl− < Br− < I− which is in agreement with our experimental findings. As can be seen in Fig. 4c (for TMAI) and Fig. 5a–c, in order to accommodate both the larger iodide and tetrafluoroborate anion and the larger tetraalkylammonium groups, the conformations of the complexes are no longer 1,3-alternate. For the TMAI, TMABF4 and TEABF4 complexes, partial-cone (“paco”) conformers are adopted and for the TBABF4 complex, a near boat-like Cs-symmetrical conformer, as shown in Fig. 3b for the calix[4]azulene host, is formed. The binding energy trends for the tetrafluoroborate salts TMABF4 > TEABF4 > TBABF4 did parallel the trend seen with the corresponding binding constants which were observed (Table 1). To possibly account for the trends in the BEs of the 1:1 complex formation in these salts, there is presumably a greater degree of entropic loss incurred in forming the complexes as the free rotations within the larger alkyl group chains become restricted within the complex.
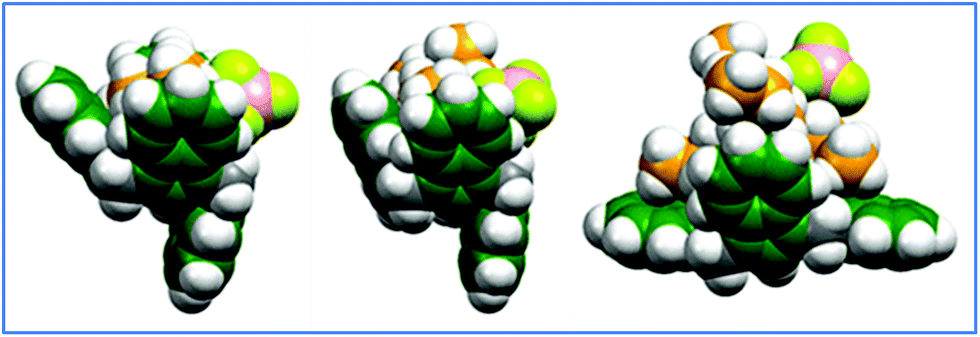 | ||
| Fig. 5 Geometry-optimized (ωB97xD/6-31G(d)) structures computed for: (a) left:TMABF4 ⊂ 1; (b) middle: TEABF4 ⊂ 1; (c) right: TBABF4 ⊂ 1. | ||
Conclusions
Calix[4]azulene 1 whose synthesis was reported by Colby and Lash in 2002 has now been shown to be a molecular receptor for tetraalkylammonium halides and thus joins the large group of cavity-containing macrocylic compounds. The binding or association constant values have been determined using the global fitting program4b with the entire UV-vis spectra derived from the titration experiments. The obtained values show trends similar to those observed by others for tetramethylammonium halides where Cl−> Br− > I−. With larger alkyl groups the trend in association constant values is Me > Et > n-Bu. DFT calculations with the dispersion-corrected, long range hybrid functional ωB97xD support these trends, while the older B3LYP density functional fails due to incorrect description of the weak inter- and intra-molecular interactions.Acknowledgements
We thank the research support from the Research Development Corporation Newfoundland and Labrador, Vale, and Memorial University of Newfoundland. The computational work has been assisted by the use of computing resources provided by WestGrid and Compute/Calcul Canada.Notes and references
- D. A. Colby and T. D. Lash, J. Org. Chem., 2002, 67, 1031–1033 CrossRef CAS PubMed.
- For leading references see: (a) Calixarenes 2001, ed. Z. Asfari, V. Böhmer, J. Harrowfield and J. Vicens, Kluwer Academic, Dordrecht, The Netherlands, 2001 Search PubMed; (b) C. D. Gutsche, Calixarenes Revisited, RSC Publishing, Cambridge, 1998 Search PubMed.
- (a) A. H Tran, D. O. Miller and P. E. Georghiou, J. Org. Chem., 2005, 70, 1115–1121 CrossRef PubMed; (b) H. F. Sleem, L. N. Dawe and P. E. Georghiou, New J. Chem., 2013, 36, 2451–2455 RSC; (c) H. F. Sleem, L. N. Dawe and P. E. Georghiou, Tetrahedron Lett., 2013, 54, 3444–3448 CrossRef CAS; (d) H. F. Sleem, L. N. Dawe, S. Rahman and P. E. Georghiou, Supramol. Chem., 2014, 26, 579–582 CrossRef CAS.
- (a) P. Thordarson, Chem. Soc. Rev., 2011, 40, 1305–1323 RSC; (b) http://www.supramolecular.org .
- X-ray structure of 1. Crystal data for calix[4]azulene: C44H32 (M = 560.69 g mol−1): monoclinic, space group C2/c (no. 15), a = 21.779(11) Å, b = 4.904(2) Å, c = 27.269(12) Å, β = 97.075(8), V = 2891(2) Å3, Z = 4, T = 153(2) K, μ(MoKα) = 0.073 mm−1, Dcalc = 1.288 g cm−3, 16217 reflections measured (5.106 ≤ 2Θ ≤ 52.984), 2982 unique (Rint = 0.0484, Rsigma = 0.0321) which were used in all calculations. The final R1 was 0.0658 (I > 2σ(I)) and wR2 was 0.1415 (all data). CCDC # 1049866.
- S. M. Bacharach, in Computational Organic Chemistry, J. Wiley & Sons, Inc., 2nd edn, 2014 Search PubMed.
- L. Fielding, Tetrahedron, 2000, 56, 6151–6170 CrossRef CAS.
- Azulene itself is known to exhibit anomalous fluorescence behaviour: (a) M. Beer and H. C. Longuet-Higgins, J. Chem. Phys., 1955, 23, 1390–1391 CrossRef CAS; (b) G. Viswanath and M. Kasha, J. Chem. Phys., 1956, 24, 574–577 CrossRef CAS and in at least one instance, as pointed out by Stella et al. (ref. 9) has led to a misinterpretation of results obtained with azulenes by Komatsu and coworkers (see ref. 10a and b).
- L. Stella, A. L. Capodilupo and M. Bietti, Chem. Commun., 2008, 4744–4746 RSC.
- (a) A. F. M. M. Rahman, S. Bhattacharya, X. Peng, T. Kimura and N. Komatsu, Chem. Commun., 2008, 1196–1198 RSC; (b) A. F. M. M. Rahman, S. Bhattacharya, X. Peng, T. Kimura and N. Komatsu, Chem. Commun., 2013, 49, 11812–11812 RSC.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- P. E. Georghiou, A. H. Tran, S. S. Stroud and D. W. Thompson, Tetrahedron, 2006, 62, 2036–2044 CrossRef CAS.
- I. Welsh and M. Lein, J. Comput. Chem., 2014, 35, 181–191 CrossRef CAS PubMed reported a DFT study on supramolecular complexation of bowl-shaped structures with fullerene C60 in which the ωB97xD functional was used with the 6-31G(d) basis set.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski, and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- H. Kruse, L. Goerigk and S. Grimme, J. Org. Chem., 2012, 77, 10824–10834 CrossRef CAS PubMed.
- G. A. Shamov, P. H. M. Budzelaar and G. Schreckenbach, J. Chem. Theory Comput., 2010, 6, 477–490 CrossRef CAS.
- G. A. Shamov, G. Schreckenbach and P. H. M. Budzelaar, J. Chem. Theory Comput., 2010, 6, 3442–3455 CrossRef CAS.
- T. Tsuneda and K. Hirao, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 375–390 CrossRef CAS.
- S. Ehrlich, J. Moellmann and S. Grimme, Acc. Chem. Res., 2013, 46, 916–926 CrossRef CAS PubMed.
- R. Sure, J. Antony and S. Grimme, J. Phys. Chem. B, 2014, 118, 3431–3440 CrossRef CAS PubMed.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724–728 CrossRef CAS.
- A. J. Fry, J. Org. Chem., 2015, 80, 3758–3765 CrossRef CAS PubMed.
- C. E. Check, T. O. Faust, J. M. Bailey, B. J. Wright, T. M. Gilbert and L. S. Sunderlin, J. Phys. Chem. A, 2001, 105, 8111 CrossRef CAS.
- (a) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 270–283 CrossRef CAS; (b) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284–298 CrossRef; (c) P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299–310 CrossRef CAS.
- (a) J. Tomasi and M. Persico, Chem. Rev., 1994, 94, 2027–2094 CrossRef CAS; (b) J. Tomasi, B. Mennucci and R. Cammi, Chem. Rev., 2005, 105, 2999–3093 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: UV-vis titration; X-ray cif and checkcif files for 1 and .mol coordinates from DFT computations. CCDC [1049866]. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra07802d |
| This journal is © The Royal Society of Chemistry 2015 |