
Continuous synthesis of a co-doped TiO2 photocatalyst and its enhanced visible light catalytic activity using a photocatalysis microreactor†
Wei Hea,
Zheng Fangb,
Kai Zhanga,
Xin Lia,
Dong Jia,
Xiubo Jiangb,
Chuanhong Qiub and
Kai Guo*a
aCollege of Biotechnology and Pharmaceutical Engineering, State Key Laboratory of Materials-Oriented Chemical Engineering, Nanjing Technology University, 30 Puzhu Rd S., Nanjing 211816, China. E-mail: hewei1989629@163.com
bSchool of Pharmaceutical Science, Nanjing Technology University, 30 Puzhu Rd S., Nanjing 211816, China. E-mail: fzcpu@163.com
First published on 17th June 2015
Abstract
Three kinds of co-doped TiO2 nanomaterials were synthesized by using a continuous precipitation method with a valve assisted micromixer in the presence of additives and titanate sources. The research results revealed that co-doped elements were incorporated into the lattice of TiO2 by substituting Ti and O atoms in the lattice of TiO2. Compared with conventional methods, the substitution process was realized within a few seconds. The resulting nanoparticles were characterized by X-ray diffraction (XRD), differential scanning calorimetry (DSC), transmission electron microscopy (TEM), selected area electron diffraction (SAED), nitrophysisorption measurements, X-ray photoelectron spectroscopy (XPS) and diffused reflectance UV-visible spectra (DRS). The synthesized samples were evaluated in the degradation of methyl orange dye (MO) under UV and visible light irradiation at room temperature. Fe, S co-doped TiO2 (TiO2–Fe–S) showed nearly the same catalytic efficiency compared with pure TiO2 (5–10 nm) under UV irradiation. The visible photocatalytic activity was higher than pure TiO2 (5–10 nm) under visible light irradiation.
1. Introduction
Recently heterogeneous semiconductor photocatalysis has become a popular technique in controlling aqueous contaminates and air pollutants. TiO2 has been an attractive material in water purification in the past decade due to its relatively low cost, strong oxidation ability, photostability and nontoxicity among various oxide semiconductors.1 More defects were observed in anatase compared with rutile,2,3 resulting in a higher photocatalytic activity. Meanwhile, higher absorptivity and adsorption rate were detected when nanoparticles with smaller particle size was used in the degradation of organic pollutants. However, decreased photocatalytic activity was observed with further smaller particle size. Recombination probability of electron–hole pairs increased when the specific surface area increased. Besides, light absorption spectral blueshifted, resulting in lower degree of photosensitization.4,5However, low photocatalytic activity under visible light was the major limitation of TiO2.6 Therefore, a great deal of efforts have been made to improve the utilization of solar energy, such as, dye sensitized,7,8 transition metals doping9–11 and noble metal deposition.12–15 More recently, TiO2 doped with multiple elements simultaneously has attracted considerable interests, since higher photocatalytic activity under visible light could be obtained. It has been reported that TiO2 doped with non-metal elements showed a relatively higher photocatalytic activity under visible light.16–18 Through mixing p orbital of nonmetal with O2p orbital of TiO2, these non-metal elements have been proved to be beneficial dopants in the TiO2 lattice to reduce the band gap energy. An optical absorption edge change from UV to visible light was observed with the doping of various transitional metal ions into TiO2.19,20 However, remarkable changes in TiO2 band gap have not been observed. Also, substitution of Ti4+ in the TiO2 lattice by metal ion, such as Fe, Ni21,22 has been reported since it could increase visible light absorbance of TiO2. Furthermore, co-doped TiO2 nanoparticles with non-metal,23–28 metal–nonmetals,29–34 metal ions,35,36 and metal–semiconductor37,38 have attracted more attention since lower band gap energy and a shift in band gap from UV into visible light have been observed.
Numerous methods have been developed to prepare and modify TiO2 nanoparticles, such as sol–gel method,39 chemical vapor depositions,40 solvothermal processes,41 sputtering,42,43 reverse micelle method,44 liquid phase deposition,45 electrochemical method46 and hydrothermal treatment.47,48 However, it is still a big challenge to control size and shape of nanoparticles, especially for nanoparticles co-doped with multiple elements. Another problem in degradation of organic water pollutants was that the degradation process was a complicated process involved in gas–liquid–solid phases.
Far-reaching progress in the development of microfluidic systems in the chemical synthesis have been obtained over the past two decades.49–51 Recently, microflow system (MFS) has been employed as a new method for preparation of nanomaterials with benefits of better control of shape and size.52 Due to extremely fast mixing and more efficient heat transfer, co-doped catalysts have been prepared by a continuous precipitation method with a valve assisted micromixer.53,54 Meanwhile, investigation concerning three-phase reaction in MFS has been launched. The effect of reactor inner diameter on space-time yield of hydrocarbon and microalgae oil conversion was studied to confirm the superiority of the microreactor for three-phase reaction.55
In this study, N–S–TiO2, B–S–TiO2 and Fe–S–TiO2 were prepared at different reaction concentrations in the MFS to increase visible light absorbance. Meanwhile, these prepared nanoparticles were employed to perform photocatalytic degradation of methyl orange (MO) dye under UV and visible light in MFS to study its potential utility in purification of organic water pollutant.
2. Experimental
2.1 Sample preparation
N–S–TiO2 catalysts were prepared in a micromixer by co-precipitation of titanium precursor solution and an alkaline solution (ammonia water, 25–28%, Sinopharm Chemical Reagent Co., Ltd). Titanous sulfate (96%, Sinopharm Chemical Reagent Co., Ltd) solution and a precipitating agent were pumped (HPLC pump, TBP 5010T, Shanghai Tauto Biotech Co., Ltd) into the valve assisted micromixer (Fig. 1). After centrifugation, white precipitates were washed with water and ethanol. Subsequently the precipitates were dried at 40 °C overnight under vacuum. Finally, N–S–TiO2 catalysts were successfully prepared by calcining white precipitates at certain temperatures for 1 h in a muffle furnace with a heating rate of 3 °C min−1. | ||
| Fig. 1 Schematic drawing of the continuous precipitation method with the valve assisted micromixer. | ||
B–S–TiO2 catalysts were prepared in the micromixer by co-precipitation of tetrabutyl titanate (98%, Sinopharm Chemical Reagent Co., Ltd) solution and a solution consisting of different concentrations of boric acid (99.8%, Sinopharm Chemical Reagent Co., Ltd) and sulfuric acid (95–98%, Sinopharm Chemical Reagent Co., Ltd). Furthermore, Fe–S–TiO2 catalysts were prepared in the micromixer by co-precipitation of tetrabutyl titanate solution and a solution including different concentrations of iron sulfate (21–23%, Sinopharm Chemical Reagent Co., Ltd).
2.2 Photocatalyst characterization
The prepared samples were characterized by differential scanning calorimetry (DSC), X-ray diffraction (XRD), transmission electron microscopy (TEM), nitrogen physisorption measurements, X-ray photoelectron spectroscopy (XPS) and diffused reflectance UV-visible spectra (DRS). The appropriate calcination temperature was determined according to the DSC results recorded in a differential thermal analysis meter (TG/DTA 7300, Exstar). XRD patterns were acquired on a Bruker D8 Advance power X-ray diffractometer with Cu Kα radiation (λ = 0.15406 nm). TEM images and SAED patterns were obtained with a JEOL JEM-200CX microscope. Nitrogen physisorption measurements were realized in a Micromeritics Asap-2020 surface area & pore size analyzer. XPS conducted using a PHI-500 VersaProbe system was employed to characterize the chemical states in the compounds. The DRS of the samples was recorded on a Perkin-Elmer Lambda-950 UV-vis spectrophotometer with an integrating sphere attachment. The extent of MO degradation was monitored using UV-vis spectrophotometer (Agilent Cary 60).2.3 Photocatalytic experiments
The photodegradation experiments were performed in a photoreactor containing an UV lamp (30 W). Better mixing between MO solution and air was obtained through the slit plate micromixer. Subsequently, the gas–liquid mixture was pumped into three-phase microreactor. The degradation experiments were performed at room temperature and pH of MO solutions was adjusted to 3.56 At given irradiation time intervals, the solutions were taken out and analyzed by an UV-vis spectrometer after centrifugation.3. Results and discussion
3.1 Optimization of flow system conditions
Table 1 showed the effects of calcination temperature and calcination time on the physical properties of TiO2–N–S powders. TiO2–N–S powders calcined at 500 °C for 1 h displayed a larger specific surface area, and its value reached 182 m2 g−1 (Table 1, entry 5). Interestingly, TiO2–N–S synthesized by sol–gel method (Table 1, entry 6) showed a lower specific area than that of TiO2–N–S prepared in the MFS. Smaller nanoparticles were obtained within few seconds in MFS, while, larger nanoparticles were prepared within several hours in the conventional method. Better mixing may be the primary cause. With increased calcination temperature and extended calcination time, the specific surface areas steadily decreased due to the growth of TiO2–N–S crystallite. It was easy to note that all powders showed a monotonic decrease in the surface areas.| Entry | Temperature (°C) | Calcination time (h) | SBETa (m2 g−1) | Crystalline sizeb (nm) | Crystalline sizef (nm) |
|---|---|---|---|---|---|
| a The BET surface area was determined by multipoint BET method.b Average crystalline size of TiO2–N–S was determined by XRD using Scherrer equation.c The samples calcined at such temperatures were amorphous particles.d The samples were prepared by a continuous precipitation method with a valve assisted micromixer.e The samples were synthesized by sol–gel method.f Average crystalline size of TiO2–N–S was determined by TEM. | |||||
| 1 | 400 | 2 | — | —c | —c |
| 2 | 500 | 2 | 178.3 | 8.589 | 8.789 |
| 3 | 600 | 2 | 104.9 | — | 10.825 |
| 4 | 500d | 1 | 182.4 | 8.189 | — |
| 5 | 500 | 0.5 | — | — | — |
| 6 | 500e | 1 | 103.8 | 10.479 | — |
| 7 | 500 | 3 | 165.7 | 9.539 | — |
The changes of phase structure of the as-prepared TiO2–N–S powder before and after heat-treatment were investigated according to the XRD results. Fig. 2 showed the effects of calcination temperature and calcination time on the phase structure of TiO2–N–S powders. The crystallinity and phase composition of the TiO2–N–S powders were obviously influenced by the calcination temperature and calcination time. With increased calcination temperatures and extended calcinations time, the intensity of anatase phase increased and the width of the (101) plane diffraction peak of anatase (2θ = 25.4°) become narrower, while, an incomplete transformation from amorphous phase to anatase phase appeared at 400 °C for 2 h. When calcined at 600 °C, two peaks were observed which may demonstrate that brookite phase was formed. The grain size of TiO2–N–S nanoparticles was estimated according to the Scherrer equation. The size of TiO2–N–S nanoparticles varied from 8.189 nm to 9.539 nm accompanying increased calcination temperatures and extended calcinations time (Table 1, entry 2, 4, 7). At the same time, co-doped nanoparticles calcined at different temperatures were also evaluated by TEM. When the nanoparticles were calcined at 400 °C, amorphous particles were observed in the TEM images (Fig. 3-a). When the calcination temperatures increased to 500 °C, the size was 8.789 nm (Fig. 3-b). However, increased temperature (600 °C) resulted in large particles (10.825 nm) (Fig. 3-c), which was in agreement with the results revealed by XRD. Therefore, the best nanoparticles were obtained when calcined at 500 °C for 1 h. Just as the trend displayed by conventional methods, specific surface area was parabolic correlated to reactant concentration in MFS.
 | ||
| Fig. 2 The XRD patterns of co-doped nanoparticles calcined at different temperatures and time. | ||
 | ||
| Fig. 3 The TEM images of co-doped nanoparticles calcined at different temperatures (a) 400 °C (b) 500 °C (c) 600 °C. | ||
3.2 DSC and XRD analysis
Fig. 4 showed the DTA-TG curves of the TiO2–N–S powders prepared in a micromixer at room temperature and dried under vacuum at 40 °C overnight. An endothermic peak at about 100 °C was due to desorption of the physically adsorbed water and residual alcohol.57 The relatively small endothermic peak at 305 °C probably come from the thermal decomposition of unhydrolyzed Ti(SO4)2 remained in the co-doped TiO2 powders.58 The DTA curve also showed two exothermic peaks at about 351 °C and 440 °C, respectively. The two peaks are probably caused by the phase transformation of amorphous to anatase.57,59 The exothermic peak at 351 °C can be assigned to the phase transformation from amorphous to anatase,60 while the other exothermic peak at a higher temperature is probably ascribed to the fact that the phase transformation of the TiO2–N–S from amorphous to anatase is suppressed by the presence of N and S elements.61 It can be concluded from the DTA results that the co-doped TiO2 sample was amorphous, and it can transform from amorphous to anatase at the appropriate temperature (351 °C, 440 °C).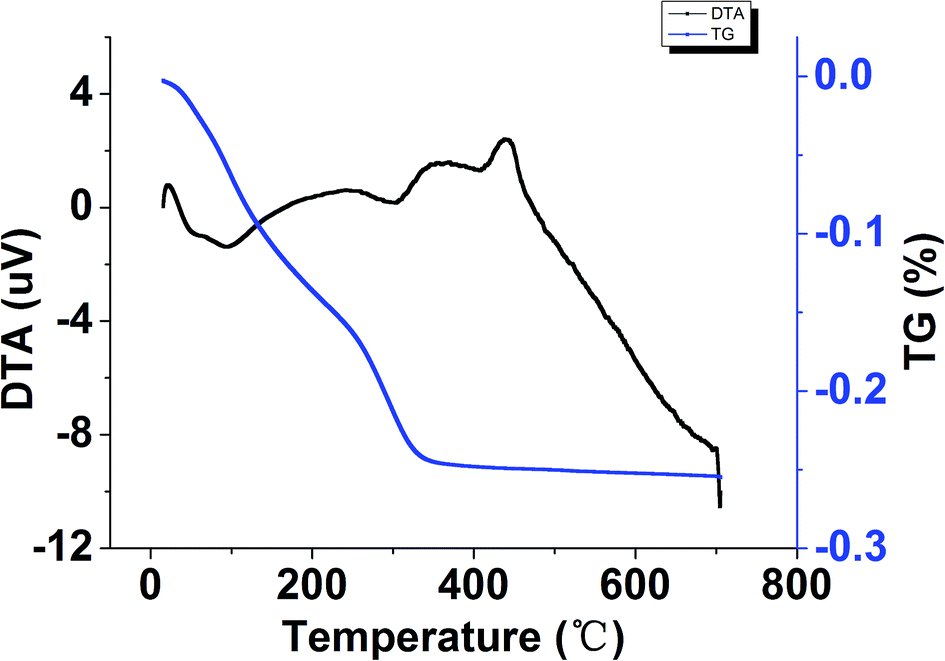 | ||
| Fig. 4 The DTA-TG curves of the TiO2–N–S powders prepared by a continuous precipitation method. | ||
3.3 HRTEM and SAED analysis
The crystallite sizes of co-doped TiO2 samples were also evaluated by HRTEM. Fig. 5 showed HRTEM images of samples in which TiO2–N–S, TiO2–B–S and TiO2–Fe–S had a crystallite sizes around 7.939, 9.321 and 8.471 nm, respectively. Fig. 5(a–c) showed the HRTEM images and SAED patterns of the TiO2–N–S, TiO2–B–S and TiO2–Fe–S nanoparticles, respectively. The doping of other elements does not leave any change in the morphology of the catalyst, which was in agreement with Hamadanian.62 Besides, a crystallite size of pure TiO2 was 10.479 nm, while, a crystallite size of Fe, S-codoped TiO2 was 8.189 nm. It can be concluded that the addition of dopants to titania hinders the growth of TiO2 nanoparticles.62 Besides, HRTEM images in Fig. 5 showed that spherical shape was the most form, while, the rest had uneven shapes. The phase structure of co-doped nanoparticles was also confirmed to be anatase phase by SAED analysis. Interestingly, the same concentric Scherrer's rings observed in the SAED images showed strong evidence for the nanocrystallinity. A set of rings instead of spots in the SAED patterns were assigned to random orientation of crystallites, which demonstrated that diffraction from different planes of nanoparticles. Meanwhile, interplanar spacings of these three nanoparticles were calculated, which were 3.42 Å, 3.46 Å and 3.51 Å, respectively. The differences between standard (101) crystal plane spacing (3.52 Å) and experiments might be attributed to structural modification caused by the co-doped elements. The location of plane corresponding to such as (101), (004), (200) and (211) was in good agreement with the Joint Committee on Powder Diffraction Standards (JCPDS no. 21-1272) reference diagrams for the corresponding anatase phase. As shown in Table 1, TiO2–N–S had a crystallite size of 10.479 nm in conventional method, while a crystallite size of 7.939 nm was obtained within few seconds in MFS. This is probably because of that fast mixing in MFS leads to fast nucleation. | ||
| Fig. 5 The HRTEM image and SAED pattern of co-doped TiO2 (a) TiO2–N–S (b) TiO2–B–S (c) TiO2–Fe–S. | ||
3.4 DRS analysis
The DRS of the samples was shown in Fig. 6. It was obvious that the absorbance of co-doped TiO2 in the visible region increased. DRS results showed a red shift in the absorption in the case of Fe–S–TiO2 that indicated that the dopant decreased band gap of the catalyst. The enhancement of absorbance in the UV-vis region increases the number of photogenerated electrons and holes to participate in the photocatalytic reaction, which can enhance the photocatalytic activity of TiO2.63 | ||
| Fig. 6 The DRS of co-doped nanoparticles prepared by a continuous precipitation method. | ||
3.5 XPS analysis
XPS analysis was performed in order to investigate the chemical composition and chemical states of TiO2 samples (Fig. 7). For pure TiO2 sample, it only contained C, O and Ti elements with sharp photoelectron peaks appearing at binding energies of 458(Ti2p), 529(O1s) and 284 eV(C1s). The C1s peak was attributed to the adventitious carbon from the XPS instrument itself. On the contrary, the TiO2–N–S samples not only contained C, O and Ti elements, but also a small amount of N and S atoms.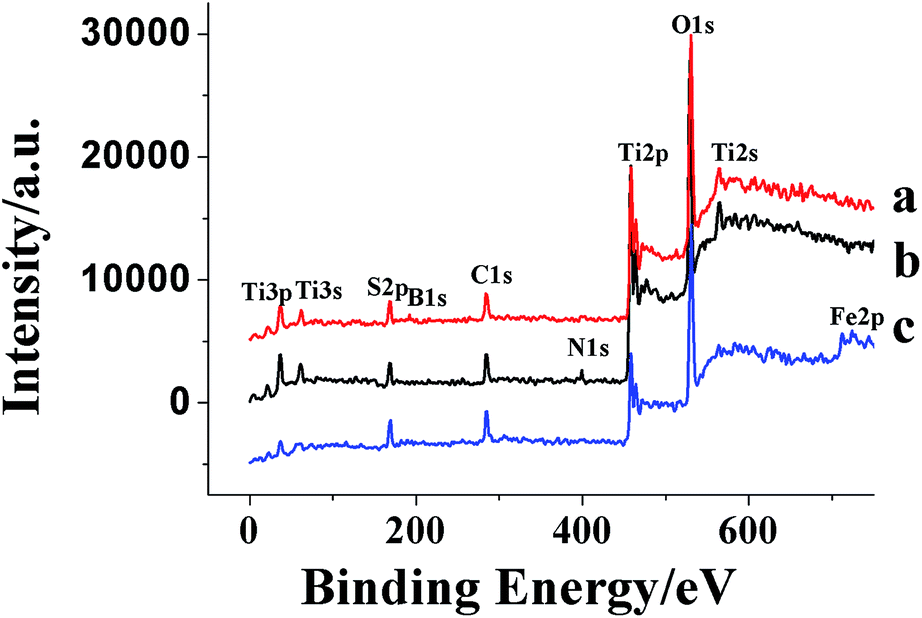 | ||
| Fig. 7 The XPS survey spectra of different co-doped TiO2. | ||
Curve b in Fig. 7 showed the high-resolution XPS spectra of the N1s region, taken on the surface of TiO2 powders. The N1s region consisted of two peaks, one was attributed to the Ti–N,64 which was probably formed by a nucleophilic substitution reaction between NH3 and Ti (SO4)2 during the hydrolysis.60 The other was probably assigned to some NH3 adsorbed on the surface of TiO2.65,66 Two S2p peaks located at 168 and 169 eV were observed. The major peak was attributed to S6+ and the peak at 169 eV was assigned to the S4+.67–70 However, it was reported that when SO2 molecules were adsorbed on the surface of TiO2, the S2p peak was in the region of 166–170 eV.71 In this study, the samples were calcined in air at 500 °C for 1 h. Under the circumstance, SO2 could not be steady absorbed on the surface of TiO2. Thus, it was more reasonable that the S4+ and S6+ were incorporated into the lattice of TiO2 and substituted for titanium atoms.
Curve a in Fig. 7 showed the XPS spectra of TiO2–B–S. A peak at 192 eV was contributed to B1s, indicating that B atoms was incorporated into the lattice of TiO2 and substituted for oxygen atoms.72 Two S2p peaks located at 168 and 169 eV were observed as well as the TiO2–N–S, indicating that the S4+ and S6+ were incorporated into the lattice of TiO2 and substituted for titanium atoms.
Curve c in Fig. 7 showed the XPS spectra of TiO2–Fe–S. Fe2p and S2p core level XPS were measured. As shown in Fig. 6, two peaks at 710.9 and 723.9 eV were assigned to Fe2p3/2 and Fe2p1/1 photoelectrons,73,74 respectively, demonstrating that Fe was incorporated into the lattice of TiO2 through substituting titanium atoms existing as Fe3+. Two S2p peaks located at 168 and 169 eV were observed as well as the TiO2–N–S, indicating that the S4+ and S6+ were incorporated into the lattice of TiO2 and substituted for titanium atoms.
3.6 Photocatalytic activity
Degradation of MO in the presence of co-doped TiO2 photocatalyst under UV and visible light irradiation was evaluated by UV-visible spectroscopy. The results were depicted in Fig. 8. Less MO was detected with increased ammonium hydroxide concentration, while the degradation level decreased when the ammonium hydroxide concentration exceeded 20 ml/100 ml. Less MO was detected with more titanous sulfate initially. However, when titanous sulfate concentration increased above 15 g/100 ml, a decrease in degradation efficiency was observed. This phenomenon indicated that the degradation process would proceed better with smaller nanoparticles. Highest degradation level was obtained when the concentration of sulfuric acid and boric acid was 1 g/100 ml. Moreover, a parabolic trend in degradation by TiO2–Fe–S was observed and the optimal degradation level was 96.92%. Mechanistically, a decrease in degradation under UV irradiation was observed with a red shift in the absorption. As shown in DRS experiments, a red shift was observed in the three co-doped TiO2. It could be seen from Fig. 9 that an increase in MO degradation by coped TiO2 was detected compared with pure TiO2 under visible light. | ||
| Fig. 8 Degradation of MO in the presence of co-doped TiO2 under UV irradiation. | ||
 | ||
| Fig. 9 Degradation of MO in the presence of photocatalysts in 3 min under visible light irradiation. | ||
3.7 Possible mechanism
Mechanistically, during the photocatalytic process, the absorption of photons by the photocatalysts leads to the excitation of electrons from the valence band to the conduction band and generates electron–hole pairs. Thus, an increase in MO degradation is observed due to the fact that S element coped in TiO2 lattice increases the surface acidity.75 Moreover, the best photocatalyst was TiO2–Fe–S, whose degradation value of 90% was obtained. With increasing Fe content, absorption in visible region and generation of electron–hole pairs increases. The high photocatalytic activity may be due to the following factors:76 (1) increased light absorption capability; (2) action of both h+/e− traps to reduce the recombination rate of h+/e− pairs during the process. Compared with TiO2–Fe–S, non-elements coped TiO2 shows a lower photocatalytic activity. This is probably because coped non-elements decreased the energy gap of TiO2, resulting in a decrease in the activity of oxidizability.77 Moreover, the lower stability of non-elements coped TiO2 may also contribute to the lower photocatalytic activity.4. Conclusions
In conclusion, a novel process for nanoparticles in MFS was investigated. Light absorption capacity increased in the visible region and an increase in MO degradation under visible light irradiation was observed with some elements co-doped TiO2 in comparison with pure TiO2. Compared with non-element coped TiO2, TiO2–Fe–S shows a higher photocatalytic activity both in UV and visible light irradiation.Acknowledgements
The research has been supported by the Joint Funds of the National Natural Science Foundation of China (Grant U1463201); National Key Basic Research Program of China (973 Program) 2012CB725204 and 2011CB710803; National High Technology Research and Development Program of China (863 Program) 2013AA031901; the National Natural Science Foundation of China (Grant no. 81302632 and 21402240); the youth in Jiangsu Province Natural Science Fund (Grant no. BK20130913); a Project Funded by the Priority Academic Program Development of Jiangsu Higher Education Institutions (PAPD).Notes and references
- A. F. Shojaie and M. H. Loghmani, Chem. Eng. J., 2010, 157, 263 CrossRef CAS PubMed.
- P. H. Maruska and A. K. Ghosh, Sol. Energy, 1978, 20, 20 CrossRef.
- K. Tanaka, M. F. V. Capule and T. Hisanaga, Chem. Phys. Lett., 1991, 187, 73 CrossRef CAS.
- X. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891 CrossRef CAS PubMed.
- A. Wold, Chem. Mater., 1993, 5, 280 CrossRef CAS.
- H. Yang, X. C. Zhang and Q. F. Tao, Inorg. Mater., 2009, 45, 1139 CrossRef CAS.
- D. F. Watson, A. Marton, A. M. Stux and G. J. Meyer, J. Phys. Chem. B, 2003, 107, 10971 CrossRef CAS.
- J. N. Clifford, E. Palomares, K. Nazeeruddin, R. Thampi, M. Grtzel and J. R. Durrant, J. Am. Chem. Soc., 2004, 125, 5670 CrossRef PubMed.
- M. Anpo and M. Takeuchi, J. Catal., 2003, 216, 505 CrossRef CAS.
- W. Zhao, C. Chen, X. Li and J. Zhao, J. Phys. Chem. B, 2002, 106, 5022 CrossRef CAS.
- G. G. Lenzi, C. V. B. Favero, L. M. S. Colpini, H. Bernable, M. L. Baesso, S. Specchia and O. A. A. Santos, Desalination, 2011, 270, 241 CrossRef CAS PubMed.
- V. Subramanian, E. E. Wolf and P. Kamat, J. Am. Chem. Soc., 2004, 126, 4943 CrossRef CAS PubMed.
- M. Jakob, H. Levanon and P. V. Kamat, Nano Lett., 2003, 3, 353 CrossRef CAS.
- C. Sahoo, A. K. Gupta and A. Pal, Desalination, 2005, 181, 91 CrossRef CAS PubMed.
- P. Wongwisate, S. Chavadej, E. Gulari, T. Sreethawong and P. Ransunvigit, Desalination, 2011, 272, 154 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269 CrossRef CAS PubMed.
- D. Chen, D. Yang, Q. Wang and Z. Jiang, Ind. Eng. Chem. Res., 2006, 45, 4110 CrossRef CAS.
- V. Iliev, D. Tomova and S. Rakovsky, Desalination, 2010, 260, 101 CrossRef CAS PubMed.
- M. I. Litter and J. A. Navio, J. Photochem. Photobiol., A, 1996, 98, 171 CrossRef CAS.
- J. C. S. Wu and C. H. Chen, J. Photochem. Photobiol., A, 2004, 163, 509 CrossRef CAS PubMed.
- X. H. Wang, J. G. Li, H. Kamiyama, Y. Moriyoshi and T. Ishigaki, J. Phys. Chem. B, 2006, 110, 6804 CrossRef CAS PubMed.
- W. Choi, A. Termin and M. R. Hoffmann, J. Phys. Chem., 1994, 98, 13669 CrossRef.
- J. Yu, M. Zhou, B. Cheng and X. Zhao, J. Mol. Catal. A: Chem., 2006, 246, 176 CrossRef CAS PubMed.
- D. Chen, Z. Jiang, J. Geng, Q. Wang and D. Yang, Ind. Eng. Chem. Res., 2007, 46, 2741 CrossRef CAS.
- X. Li, R. Xiong and G. Wei, Catal. Lett., 2008, 125, 104 CrossRef CAS.
- T. Ohno, T. Tsubota, Y. Nakamura and K. Sayama, Appl. Catal., A, 2005, 288, 74 CrossRef CAS PubMed.
- L. Lin, R. Y. Zheng, J. L. Xie, Y. X. Zhu and Y. C. Xie, Appl. Catal., B, 2007, 76, 196 CrossRef CAS PubMed.
- F. Wei, L. Ni and P. Cui, J. Hazard. Mater., 2008, 156, 135 CrossRef CAS PubMed.
- C. Liu, X. Tang, C. Mo and Z. Qiang, J. Solid State Chem., 2008, 181, 913 CrossRef CAS PubMed.
- P. Wu, J. Tang and D. Zhi, Mater. Chem. Phys., 2007, 103, 264 CrossRef CAS PubMed.
- H. Xia, H. Zhuang, D. Xiao and T. Zhang, J. Wuhan Univ. Technol., Mater. Sci. Ed., 2008, 23, 467 CrossRef CAS PubMed.
- D. B. Hamal and K. J. Klabunde, J. Colloid Interface Sci., 2007, 311, 514 CrossRef CAS PubMed.
- K. Obata, H. Irie and K. Hashimoto, Chem. Phys., 2007, 339, 124 CrossRef CAS PubMed.
- X. Yang, C. Cao, K. Hohn, L. Erickson, R. Maghrang, D. Hamal and K. Klabunde, J. Catal., 2007, 252, 296 CrossRef CAS PubMed.
- D. R. Zhang, Y. H. Kim and Y. S. Kang, J. Curr. Appl. Phys., 2006, 6, 801 CrossRef PubMed.
- R. Khan, S. W. Kim, T. Kim and C. Nam, Mater. Chem. Phys., 2008, 112, 167 CrossRef CAS PubMed.
- X. Yang, L. Xu, X. Yu and Y. Guo, Catal. Commun., 2008, 9, 913 CrossRef PubMed.
- B. Zhao, B. Shi, X. Zhang, X. Cao and Y. Zhang, Desalination, 2011, 268, 55 CrossRef CAS PubMed.
- K. Abbas, Colloids Surf., A, 2009, 346, 130 CrossRef PubMed.
- W. L. Gladfelter, Surf. Sci., 2011, 605, 1146 CrossRef CAS PubMed.
- S. Perera and E. G. Gillan, Solid State Sci., 2008, 10, 864 CrossRef CAS PubMed.
- Y. Hoshi, D. Ishihara, T. Sakai, O. Kamiya and H. Lei, Vacuum, 2010, 84, 1377 CrossRef CAS PubMed.
- X. Yu and Z. Shen, Vacuum, 2011, 85, 1026 CrossRef CAS PubMed.
- X. Sui, Y. Chu, S. Xing and C. Liu, Mater. Lett., 2004, 58, 1255 CrossRef CAS PubMed.
- M. Mallak, M. Bockmeyer and P. Labmann, Thin Solid Films, 2007, 515, 8072 CrossRef CAS PubMed.
- N. Lu, H. Zhao, J. Li, X. Quan and S. Chen, Sep. Purif. Technol., 2008, 62, 668–673 CrossRef CAS PubMed.
- T. Putta, M. C. Lu and J. Anotai, J. Environ. Manage., 2011, 92, 2272 CrossRef CAS PubMed.
- S. S. Mali, P. S. Shinde, C. A. Betty, P. N. Bhosale, W. J. Lee and P. S. Patil, Appl. Surf. Sci., 2011, 257, 9737 CrossRef CAS PubMed.
- K. S. Elvira, X. C. Solvas, R. C. R. Wootton and A. J. deMello, Nat. Chem., 2013, 5, 905 CrossRef CAS PubMed.
- B. Xu, Y. Zhang, S. Wei, H. Ding and H. Sun, ChemCatChem, 2013, 5, 2091 CrossRef CAS PubMed.
- N. Wang, X. Zhang, Y. Wang, W. Yu and H. L. W. Chan, Lab Chip, 2014, 14, 1074 RSC.
- G. Simson, E. Prasetyo, S. Reiner and O. Hinrichsen, Appl. Catal., A, 2013, 450, 1 CrossRef CAS PubMed.
- Scientific Bases for the Preparation of Heterogeneous Catalysts, ed. E. M. Gaigneaux, M. Devillers, D. E. DeVos, S. Hermans, P. A. Jacobs, J. A. Martens and P. Ruiz, Elsevier, 2006, p. 175 Search PubMed.
- S. Kaluza and M. Muhler, J. Mater. Chem., 2009, 19, 3914 RSC.
- L. Zhou and A. Lawal, Energy Fuels, 2015, 29, 262 CrossRef CAS.
- L. Rizzo, J. Koch, V. Belgiorno and M. A. Anderson, Desalination, 2007, 211, 1 CrossRef CAS PubMed.
- J. Yu, X. Zhao and Q. Zhao, Thin Solid Films, 2000, 379, 7 CrossRef CAS.
- J. C. Yu, J. G. Yu, W. K. Ho, Z. T. Jiang and L. Z. Zhang, Chem. Mater., 2002, 14, 3808 CrossRef CAS.
- K. Terabe, K. Kato, H. Miyazaki, S. Yamaguchi, A. Imai and Y. Iguchi, J. Mater. Sci., 1994, 29, 1617 CrossRef CAS.
- J. Yu, J. C. Yu, W. Ho, M. K. P. Leung, B. Cheng, G. Zhang and X. Zhao, Appl. Catal., A, 2003, 255, 309 CrossRef CAS.
- J. Yu, M. Zhou, B. Cheng and X. Zhao, J. Mol. Catal. A: Chem., 2006, 246, 176 CrossRef CAS PubMed.
- M. Hamadanian, A. Reisi-Vanani, M. Behpour and A. S. Esmaeily, Desalination, 2011, 281, 319 CrossRef CAS PubMed.
- D. Li, H. Haneda, S. Hishita and N. Ohashi, Mater. Sci. Eng. B, 2005, 117, 67 CrossRef PubMed.
- S. Sakthivel, M. Janczarek and H. Kisch, J. Phys. Chem. B, 2004, 108, 19384 CrossRef CAS.
- O. Diwald, T. L. Thompson, E. Zubkov, E. G. Goralski, S. D. Walck Jr and J. T. Yates, J. Phys. Chem. B, 2004, 108, 6004 CrossRef CAS.
- R. Nakamura, T. Tanaka and Y. Nakato, J. Phys. Chem. B, 2004, 108, 10617 CrossRef CAS.
- T. Ohno, T. Mitsui and M. Matsumura, Chem. Lett., 2003, 32, 364 CrossRef CAS.
- M. Zhou and J. Yu, J. Hazard. Mater., 2008, 152, 1229 CrossRef CAS PubMed.
- S. Liu and X. Chen, J. Hazard. Mater., 2008, 152, 48 CrossRef CAS PubMed.
- T. Ohno, T. Tsubota, K. Nishijima and Z. Miyamoto, Chem. Lett., 2004, 33, 750 CrossRef CAS.
- D. I. Sayago, P. Serrano, O. Bohme, A. Goldoni, G. Paolucci, E. Roman and J. A. Martin-Gago, Phys. Rev. B, 2001, 64, 205402 CrossRef.
- B. Wang, X. Lu, J. Xuan and M. K. H. Leung, Mater. Lett., 2014, 115, 57 CrossRef CAS PubMed.
- T. K. Ghorai, S. K. Biswas and P. Pramanik, Appl. Surf. Sci., 2008, 254, 7498 CrossRef CAS PubMed.
- M. Descostes, F. Mercier, N. Thromat, C. Beaucaire and M. Gautier-Soyer, Appl. Surf. Sci., 2000, 165, 288 CrossRef CAS.
- T. Ohno, M. Akiyoshi, T. Umebayashi, K. Asai, T. Mitsui and M. Matsumura, Appl. Catal., A, 2004, 265, 115 CrossRef CAS PubMed.
- M. Hamadanian, A. Reisi-Vanani, M. Behpour and A. S. Esmaeily, Desalination, 2011, 281, 319 CrossRef CAS PubMed.
- R. Nakamura, T. Tanaka and Y. Nakato, J. Phys. Chem. B, 2004, 108, 10617 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05956a |
| This journal is © The Royal Society of Chemistry 2015 |