
Kinetic study and peak purity determination of bupropion hydrochloride using RRLC/DAD and HPLC/MWD methods: stability study and application in pharmaceutical preparation and in synthetic mixtures with nicotine†
Abstract
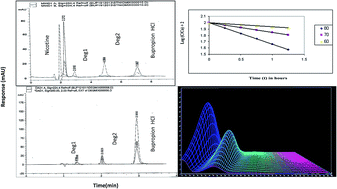
Two simple, specific, and precise stability indicating chromatographic methods have been developed, optimized, and validated for the purity determination of bupropion HCl (BUP) in bulk powders, pharmaceutical preparations as well as in the presence of its hydrolytic and oxidative degradation products and its co-administered drug nicotine (NIC). The first method was based on the determination of the cited drug using High Performance Liquid Chromatography (HPLC). The adequate separation of BUP from its degradation products and NIC was obtained using an Intertsil ODS3 (250 × 4.6 mm i.d., 5 μm particle size) column. The method was used to investigate the kinetics of the alkaline and oxidative degradation processes of BUP where the order rate constants, half lives, and activation energies were calculated. The second method was based on using Rapid Resolution Liquid Chromatography (RRLC) to separate BUP from its degradation products on a XDB C18 (50 × 4.6 mm i.d., 1.8 μm particle size) column. The linear ranges were 5–100 and 2–20 μg mL−1 with LOD of 1.33 and 0.2 μg mL−1 for the HPLC and RRLC methods, respectively. The quantification in both methods was based on coupling the separation with dual wavelength detection at 250 nm for BUP and NIC and at 224 nm for the degradation products. The peak purity of BUP in its pharmaceutical preparation spiked with its degradates revealed symmetry factors (999.935 & 999.963) within the calculated thresholds (>999.841 & >999.901) for HPLC and RRLC, respectively. The suggested methods were validated in compliance with the ICH and USP guidelines. The assay methods were successfully used to estimate BUP in Wellbutrin® 150 SR tablets and good percentage recoveries were obtained. The developed methods were statistically compared with the official USP methods and compared favourably with no significant difference in terms of accuracy and precision.
 Please wait while we load your content...
Please wait while we load your content...

