
The Biginelli reaction under batch and continuous flow conditions: catalysis, mechanism and antitumoral activity†
Gabriel C. O. Silvaa,
Jose R. Correab,
Marcelo O. Rodrigues*a,
Haline G. O. Alvimb,
Bruna C. Guidob,
Claudia C. Gattob,
Kaline A. Wanderleya,
Mariana Fioramontec,
Fabio C. Gozzoc,
Rodrigo O. M. A. de Souzad and
Brenno A. D. Neto*b
aLIMA – Laboratório de Inorgânica e Materiais, Campus Universitário Darcy Ribeiro, CEP 70904970, P.O. Box 4478, Brasilia, DF, Brazil. E-mail: marcelozohio@unb.br
bLaboratory of Medicinal and Technological Chemistry, University of Brasilia (IQ-UnB), Campus Universitario Darcy Ribeiro, CEP 70904970, P.O. Box 4478, Brasilia, DF, Brazil. E-mail: brenno.ipi@gmail.com
cInstitute of Chemistry, University of Campinas (Unicamp), Campinas, São Paulo 13083-970, Brazil
dBiocatalysis and Organic Synthesis Group, Chemistry Institute, Federal University of Rio de Janeiro, CEP 21941909, Rio de Janeiro-RJ, Brazil
First published on 19th May 2015
Abstract
Two novel coordination polymers (CPs) have been synthesized, characterized and successfully applied as robust heterogeneous catalysts for the Biginelli multicomponent reaction to obtain 3,4-dihydropyrimidin-2(1H)-one or thione (DHPMs) derivatives. The reaction was initially developed using both CPs and the Zn-based material showed much better catalytic activity. After the reaction optimization under batch conditions, a continuous flow protocol was developed and applied with impressive results. Four bioactive DHPMs were successfully synthesized with high yields. The mechanism of the transformation was also investigated by electrospray (tandem) mass spectrometry (ESI-MS(/MS)) analyses. Online monitoring of the reaction indicated under the developed conditions that the iminium mechanism is preferred over the enamine- and Knoevenagel-based mechanisms. Nine DHPMs had their antitumoral activities evaluated against MCF-7 (human breast cancer cells), A549 (human alveolar basal epithelial cells) and Caco-2 (human epithelial colorectal cells) cancer cell lineages. Fibroblasts (healthy cells) were not affected by the tested DHPMs showing an excellent selectivity for tumour cells. Three DHPMs returned impressive results, being capable of inhibiting tumour cell proliferation in 72 h.
Introduction
Coordination polymers (CPs) and metal–organic frameworks (MOFs), which are a class and a subclass of hybrid materials, are among the most iconic compounds investigated in the last two decades.1 Initially, several research groups devoted massive efforts to producing new CPs and MOFs with the most diverse types of organic ligands and metal ions. This effort aimed at understanding the complex algorithms responsible for the myriad of possibilities for framework constructions and for their intriguing topological features. The net result of these early studies was considerable progress concerning theoretical and practical approaches of controlled syntheses of novel CPs and MOFs. In this regard, the judicious choices of both organic ligands and metal ions, as well as controlled synthetic conditions (stoichiometry, temperature, pH, time, etc.), may be considered the crucial elements which define the structural and functional aspects of a new CP or MOF.1,2More recently, MOFs and CPs have been seen as hybrid materials at the interface between synthetic chemistry and material sciences, being also considered of paramount importance for biomedical,3 sensing,4 gas storage,5 separation6 and catalytic application.7–9 Taking into account the possibilities of never-ending designs (metal–ligand combinations) and crystal engineering,10 it is reasonable to envisage CP/MOF properties being finely tuned towards appropriate size, shape, topology, dimensionality, and chemical environment11 aiming at an efficient catalytic transformation.12 Indeed, since the landmark publication of Ogura and coworkers (ten years ago) describing the first catalytic application of a MOF structure for the cyanosilylation reaction of aldehydes,13 many achievements have been observed in the catalysis field and catalytic application of such materials.14–16 Even so, the potential of CPs/MOFs for catalytic applications is only beginning to be exploited. To some extent, this is because of their relative thermal and hydrolytic instability when compared to well-established zeolites. The engineering of strong Lewis acid or Bronsted acid sites in CPs/MOFs still remains a great challenge to be overcome.9 Beyond these limitations, it is possible to add their inherent high sensitivity to moisture and air instability.17 Therefore, a successful application of new CP/MOF scaffolds lies in the robustness and stability of the new material which is inwardly associated with its proper design and synthesis.
Multicomponent reactions (MCRs) are today unsurpassed synthetic tools for the production of bioactive structure libraries.18 Considering that most of the atoms of the starting materials (three or more components) are incorporated into the final product, and that water is the sole byproduct for some MCRs, these reactions became synthetic tools found in a prominent position. In this context, many catalytic methodologies have been tested to improve MCR adduct formation, to reduce reaction times, to improve yields, selectivities and for more amenable reaction conditions, as noted in many recent reviews.19–23 Among MCRs, the Biginelli reaction (Scheme 1), announced by Pietro Biginelli in 1891,24 is a very elegant multicomponent methodology applied in the synthesis of 3,4-dihydropyrimidin-2(1H)-one or thione (DHPMs) derivatives. DHPMs usually have distinct biological activities and can be applied as calcium channel modulators, adrenergic receptor antagonists, antibacterials, mitotic Kinesin inhibitors, antivirals, and others, as reviewed elsewhere.25–27 Importantly, DHPMs typically have pronounced biological activities in their racemic forms,28 therefore increasing the interest in direct and efficient methodologies for their synthesis.
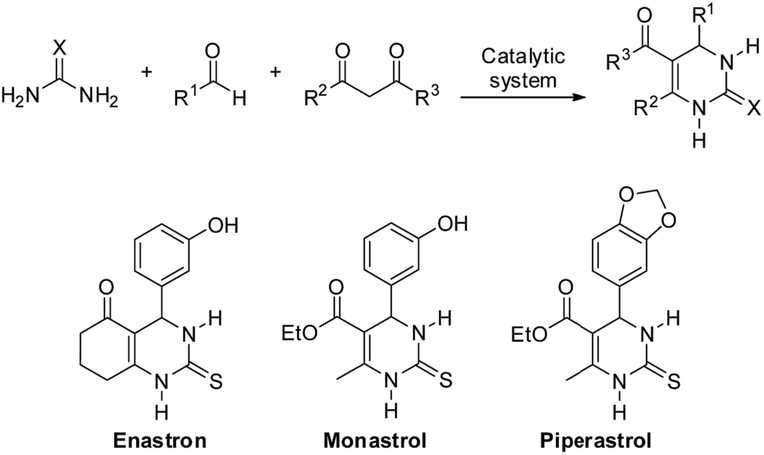 | ||
| Scheme 1 The general Biginelli reaction applied to the synthesis of 3,4-dihydropyrimidin-2(1H)-one or thione (DHPM) derivatives (top) and examples of known bioactive DHPMs (bottom). | ||
In this context, the courtship between CPs/MOFs and MCRs has produced promising results which opened up a large but as yet unexploited avenue of possibilities. Some MCRs, for instance, have been recently performed catalysed by CPs/MOF scaffolds with relative successes;29–31 therefore fostering interest in the development of new stable and more robust hybrid materials capable of performing catalysed MCRs. Two coordination polymer32,33 scaffolds have already been used as catalysts for the Biginelli reaction; and these two examples may be considered pioneering works on the use of such hybrid materials for the Biginelli MCR. However, the application of robust new CPs/MOFs as heterogeneous catalysts for the Biginelli reaction still needs to be tested. Very recently,34 a MOF scaffold was tested as the heterogeneous catalyst for the Biginelli reaction with relative success, but the tested material was already known for its instability, especially in the presence of moisture,35–37 which is a highly limiting feature. Most motivated by our interest in MCRs,38 especially in the Biginelli reaction,39–41 and due to our expertise in the synthesis and application of CPs/MOFs,42–44 we describe herein a successful strategy for combining novel robust (stable) CPs with the Biginelli MCR. In the current manuscript, we describe the synthesis, characterization, photophysical properties and catalytic application of two novel stable and robust zinc- and cadmium-based CPs as the heterogeneous catalysts for the Biginelli reaction, with impressive results under batch and continuous flow conditions. The mechanism of the transformation, as well as the antitumoral activities of the synthesized DHPMs, are also evaluated and disclosed herein.
Results and discussion
Two new CPs were obtained via hydrothermal synthesis and isolated as well-formed single crystals suitable for single crystal X-ray analyses (Fig. 1 and 2). Table 1 shows the details of the structure solutions and final refinement for CP 1 and CP 2. The X-ray powder patterns for 1 and 2 were also consistent with the structural models, as can be observed in the final Rietveld plots (see Fig. S1 in the ESI†). | ||
| Fig. 1 (a) Asymmetric unit of CP 1. (b) Schematic representation of distorted tetrahedron coordination environment of the Zn2+ ion. Symmetry transformation used to generate equivalent atoms: I x, 1.5 − y, −0.5 + z; II 1 − x, 1 − y, 1 − z; III x, 1 + y, z; IV x, 0.5 − y,−0.5 + z. (c)–(e) Views along a, b and c axis of the extended structure of 1. | ||
 | ||
| Fig. 2 (a) Asymmetric unit of CP 2. (b) Schematic representation of distorted trigonal prism monocapped coordination environment of the Cd2+ ion. Symmetry transformation used to generate equivalent atoms: I −0.5 + x, 0.5 − y, z; II 0.5 − x, 0.5 + y, 1 − z. (c) and (d) Views along a and c axes of the extended structure of 2. | ||
| Identification code | (1) | (2) |
| Chemical formula | C8H2Cl2O4Zn | C8H2Cl2O4Cd |
| M (g mol−1) | 298.37 | 345.40 |
| Crystal system | Monoclinic | Monoclinic |
| Space group | P21/c | P21/a |
| Unit cell a (Å) | 16.991(4) | 7.882(5) |
| b (Å) | 7.158(2) | 7.035(5) |
| c (Å) | 7.480(2) | 16.863(5) |
| β | 99.014(10) | 97.547(5) |
| V (Å3) | 898.48(4) | 927.0(9) |
| Z | 4 | 4 |
| Dc/g cm−3 | 2.206 | 2.475 |
| Index ranges | −24 ≤ h ≤ 24 | −8 ≤ h ≤ 9 |
| −10 ≤ k ≤ 10 | −7 ≤ k ≤ 8 | |
| −10 ≤ l ≤ 10 | −16 ≤ l ≤ 20 | |
| Absorption coefficient/mm−1 | 3.312 | 2.917 |
| Absorption correction | Semi-empirical from equivalents | Semi-empirical from equivalents |
| Max/min transmission | 0.724/0.269 | 0.745/0.628 |
| Measured reflections | 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 695 695 |
4523 |
| Independent reflections/Rint | 2885/0.0271 | 1645/0.0155 |
| Refined parameters | 145 | 136 |
| R1 (F)/wR2 (F2) (I > 2σ(I)) | 0.0247/0.0578 | 0.0227/0.0573 |
| GooF | 1.040 | 1.125 |
| Largest diff. peak and hole (eÅ−3) | 0.560 and −0.495 | 0.909 and −0.432 |
| Deposit number CCDC | 1020344 | 1020345 |
Single crystal analysis revealed that CP 1 crystallizes in a monoclinic system with the space group of P21/c. As depicted in Fig. 1, the asymmetric unit of 1 is composed of one crystallographically independent Zn2+ cation, which is coordinated with one DCPHA (4,5-dichlorophthalic acid) ligand. Each Zn2+ center is inserted in a distorted trigonal bipyramidal environment by O1, O1I, O2II, O3III and O4IV atoms from carboxylate groups of four distinct DCPHA anions. The O1–Cu–O1′ angles between the zinc ion and apical coordinated oxygen atoms is 173.79(5)° and the base of the trigonal bipyramid display bond angles near of 120° in the range of 111.71(6)° and 126.31(5)°.
The Zn–O length is in the range of 1.925(11)–2.406(11) Å, which is in close agreement with those found in related Zn CPs with aromatic carboxylate ligands.45 In CP 1, each DCPHA ligand is coordinated to five Zn2+ ions via two coordination modes typically observed in –COO− groups: one carboxylate group is coordinated with three Zn2+ ions via μ2-η1:η1-syn–syn-bridging mode, whereas the other one is bonded to the remaining metal cations through a μ3-η2:η1 chelating/bridging coordination mode. The highly distorted trigonal bipyramidal geometry, ZnO5, shares the edges with two others via the symmetrically related O1 atoms, resulting in an infinite 1D running along the c axis, which is further extend by distinct DCPHA ligands along the b and c axes to result in a 2D layered framework (Fig. 1(c)–(e)). The Zn⋯Zn distances across the chains are in the range of 3.750–4.406 Å.
The X-ray data showed that the structure of CP 2 is an extended 2D framework, which crystallizes in a monoclinic system with the space group of P21/a. The asymmetric unit of 2 is constituted by one crystallographically independent Cd2+ ion coordinated with one DCPHA ligand (see Fig. 2(a)). The DCPHA anion is linked to four metal centers via μ3-η2:η2 and μ2-η2:η1 chelating/bridging coordination modes. The Cd2+ ion is coordinated by O4, O4I, O5I, O5II, O6, O6II and O7I oxygen atoms from four organic ligands, so the chemical environment is best described as a distorted monocapped trigonal prism. In Fig. 2(b) the CdO7 polyhedra are gripped by DCPHA ligands to form a 2D architecture, as represented in Fig. 2(c)–(d). The Cd⋯Cd distances across the chains are in the range of 3.86–4.382 Å. The Cd–O bond lengths vary from 2.260(3) to 2.510(3) Å and the O–Cd–O bond angles are in the range of 53.57(8)–153.23(8)°.
To prove the water stability of the new materials, we added water (1 mL mg−1) and heated the mixture for four hours at 100 °C. The water was removed and the materials dried. Both recovered CPs had the same crystalline phases as the originals. After catalysis and recycle, new powder X-ray analysis were performed and are discussed in due course.
It is well-established that aromatic polycarboxylate ligands play an important role in the production and design of novel CPs/MOFs, due to their rigidity and flexibility in coordination mode that provide the construction of the one-dimensional chains until 3D architectures. The substitute halogen for aromatic rings has an important influence on the spectroscopic properties of these organic ligands. In halogen-substituted benzenedicarboxylate ligands a decrease in fluorescence is normally observed as a function of the atomic height of the halogen with a subsequent increase in the phosphorescence. The probability of the singlet to triplet intersystem crossing is enhanced due to the heavy atom effect. Fig. 3 exhibits a comparison between the emission spectra of 1, 2 and DCPHA ligand acquired at room temperature.
 | ||
| Fig. 3 Emission spectra acquired at room temperature upon excitation at 330 nm. The blue, red and black circles indicate 1, 2 and DCPHA ligand, respectively. The inset shows the CIE chromatic diagram for 1, 2 and DCPHA. | ||
The free ligand, 1 and 2 exhibit board bands in the spectral range from 370–900 nm (λexc = 330 nm) with the maxima centered at 450 (2222 cm−1), 522 (19157 cm−1), and 470 nm (21276 cm−1), respectively. It is possible to correlate the position of the emission band with the dihedral angle of the aromatic rings in CP structures.46,47 Small dihedral angles result in high π–π interaction, which shifts the emission bands to lower energies, whereas large dihedral angles lead to low interaction of the π electrons therefore shifting the emission band to higher energies.48,49 In accordance with the crystallographic data, the dihedral angles (θ) of the aromatic rings for 1 and 2 are 4.380° and 27.360°, respectively. These results are in excellent agreement with the red-shift of 3065 cm−1 and 946 cm−1 observed for structures 1 and 2 when compared with the free ligand. The CIE chromaticity diagram shows that the differences between the emission bands of 1, 2 and DCPHA ligand are reflected in their color coordinates (1: x = 0.29, y = 0.44; 2: x = 0.23, y = 0.39; DCPHA: x = 0.19, y = 0.29).
After the material characterizations, the two CPs were applied as the heterogeneous catalysts for the Biginelli reaction. The Biginelli reaction is a three-component MCR which allows direct access to a 3,4-dihydropyrimidin-2(1H)-one (or thione) known as a DHPM.50 Some DHPMs directly synthesized using the Biginelli reaction are bioactive compounds25 which can be used in their racemic form, sustaining their biological activity.51–53 To this end, many new catalytic systems, especially new materials, have been very recently described aiming at improving DHPM syntheses.54–64
In this context, the new materials were tested as heterogeneous catalysts for the Biginelli MCR. The model reaction (i.e. a mixture of 1.00 mmol of benzaldehyde, 1.00 mmol of ethyl acetoacetate and 1.00 mmol of urea) was conducted under solvent-free conditions in order to avoid the use of any organic solvent. Solvent-free conditions for MCRs are a trend65–67 and are in line with the green features of the Biginelli reaction. The catalytic tests conducted using 2 as the catalyst showed poor activity for the Biginelli reaction, whereas the use of 1 showed promising results and, in the first reaction hour at 100 °C, the Biginelli adduct was obtained in 25% using only 1 mol% of the catalyst. Considering that Cd is a toxic metal, the use of the Zn-based CP (1) is by far more attractive from the viewpoint of ecology and green chemistry. For this reason, we decided to maintain the catalytic tests only with the 1 and only in solvent-free versions, which is considered to be a benign pathway to sustainability.68 The reaction profile (reaction time) and catalyst amount were evaluated and the results are better visualized in Fig. 4.
 | ||
| Fig. 4 Reaction profile (left) and catalyst concentration (right) to optimize the catalytic system performance. The experiments shown on the left were conducted with only 1 mol% of the catalyst (1). The experiments shown on the right refer to 5 hours of reaction. All reactions were conducted at 100 °C with equimolar quantities of the three reagents under solvent-free conditions and with 1 as the heterogeneous catalyst. | ||
Most Biginelli reports describe the optimized conditions for new catalytic systems using at least one of the reagents in excess (see the cited reviews). In the current work, the catalytic system was optimized for the use of equimolar quantities of the reagents (no reagent excess at all). As noted in the reviews, only a few reports describe the use of equimolar quantities of the three reagents and, in this sense, the current work is one of these few exceptions. Reaction conditions using 1–4 mol% already returned satisfactory results, but considering that the substrate variation is usually more difficult to conduct for the Biginelli reaction, we decided to use 5 mol% as the catalyst amount in the sequence of the reactions. Likewise, Fig. 4 depicts the catalyst-free version, which afforded the desired DHPM in only 51%; this was therefore a very limited yield with a huge waste of the starting materials, which is found in contrast with a expected green system for the Biginelli reaction. After the optimization of the catalytic conditions, several DHPMs were also synthesized and the results are summarized in Table 2.
|
|||||
|---|---|---|---|---|---|
| DHPM (4a–j) | Product | Yield (%) | |||
| R1 | R2 | R3 | X | ||
| 4a | Ph | CH3 | CH3CH2O | O | 91 |
| 4b | Ph | CH3 | CH3CH2O | S | 87 |
| 4c | 3-OH-Ph | CH3 | CH3CH2O | O | 90 |
| 4d | 3-OH-Ph | CH3 | CH3CH2O | S | 92 |
| 4e | 3-NO2-Ph | CH3 | CH3CH2O | O | 95 |
| 4f | H | CH3 | CH3CH2O | O | 99 |
| 4g | 4-OH-3-OCH3-Ph | CH3 | CH3CH2O | O | 80 |
| 4h | 3-OH-Ph |  |
S | 87 | |
| 4i | 3-OH-Ph | 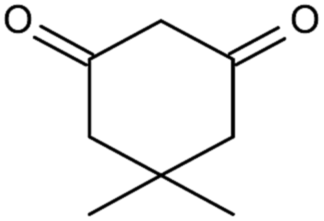 |
S | 91 | |
| 4j | 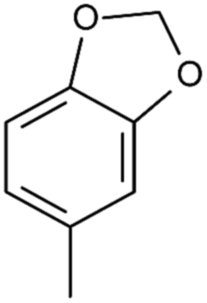 |
CH3 | CH3CH2O | S | 83 |

It is notable that the known bioactive compounds Oxomonastrol (Table 2, 4c) Monastrol (Table 2, 4d), Enastron (Table 2, 4h), Dimethylenastron (Table 2, 4i) and Piperastrol (Table 2, 4j) were obtained in a one-pot version with high yields with no reagent excess in only 5 hours.
The use of a heterogeneous catalyst pointed to an efficient recycle of the catalytic system. After the completion of the reactions, the products were extracted with hot DMSO or ethanol and the catalyst was centrifuged, filtrated and used at least three times without efficiency loss (98% for both the second and the third runs) or structural collapse (Fig. S1†). Moreover, the product solution collected at the end of the catalytic cycles were analysed by ESI(+)-MS(/MS) and none Zn2+ was detected, which confirms the non-leaching of the heterogeneous metal catalyst. After three runs, however, we noticed the structural collapse (see Fig. S1†). Most of the synthesized DHPMs precipitate in the reaction medium thus making the separation of the Biginelli adduct and the catalyst a laborious task, but possible, as noted. At this point, we envisaged the possibility of a more efficient process aiming to overcome this drawback and considering the possibility of scale-up. The inherent feature of the catalyst (heterogeneous and insoluble in most solvents) renders 1 an attractive material for use as a heterogeneous catalyst under continuous flow conditions. Therefore, after achieving these excellent results for the Biginelli reaction under batch conditions, we decided to optimize the reaction settings in order to develop a continuous flow protocol. Much attention has been given to continuous flow reactions in recent years; and the development of a heterogeneous catalysis protocol using packed bed reactors is a growing field of high interest for the chemical community. Continuous process technology is indeed regarded as a tool for sustainable production, as very recently reviewed.69 A promising continuous flow condition for the Biginelli reaction has already been described by Kappe and coworkers.70 This landmark publication70 highlighted the potential of the strategy for the Biginelli MCR, and a later review by Kappe and Glasnov,71 pointed to the importance of new and efficient catalysts for this transformation under continuous flow conditions and for other heterocyclic syntheses.
In this context, we have started optimizing the model reaction for a continuous flow protocol using CP 1 as the heterogeneous catalyst. Under continuous flow conditions, we could not run a solvent-free version of the reaction since urea was not completely soluble in the reaction mixture. Among the tested organic solvents, DMF proved to be the best choice. The reaction mixture in DMF was then pumped through the packed bed reactor, filled with catalyst 1, at different temperatures and flow rates, in order to verify the influence of each variable on reaction yields, as shown in Table 3.

The results presented in Table 3 show that the continuous flow protocol can be very effective in reducing the reaction time from hours to minutes. Temperature has an important effect, since at higher temperatures (100 °C in this case), higher flow rates can be used in order to achieve higher yields in shorter reaction times (Table 3, entry 2). Additionally to these important features, the catalyst could be efficiently recycled at least three times (99%, 97% and 98%, respectively) without any notable change in the reaction outcome and without the laborious separation process of the catalyst and the Biginelli adduct.
Success in developing this strategy led us to expand the scope of the continuous flow protocol (DMF as solvent, 100 °C and flow rate of 0.1 mL min−1) to the direct synthesis of other DHPMs (Table 4), especially for bioactive derivatives.
|
|||||
|---|---|---|---|---|---|
| DHPM | Product | Yield (%) | |||
| R1 | R2 | R3 | X | ||
| a Packed bed reactor filled with 250 mg of catalyst 1 (50 mm × 6.6 mm). | |||||
| 4b | Ph | CH3 | CH3CH2O | S | >99 |
| 4c | 3-OH-Ph | CH3 | CH3CH2O | O | 92 |
| 4d | 3-OH-Ph | CH3 | CH3CH2O | S | 92 |
| 4j |  |
CH3 | CH3CH2O | S | 82 |

As shown in Table 4, a variety of DHPMs could be synthesized using the continuous flow protocol with similar yields to those obtained under batch conditions but with reduced reaction time and with the possibility of recycling (as already demonstrated). The bioactive derivatives known as Oxomonastrol (4c), Monastrol (4d) and Piperastrol (4j) were directly obtained in high yields (typically above 90%).
The mechanism of the Biginelli reaction promoted by catalyst 1 has been investigated by electrospray (tandem) mass spectrometry – ESI-MS(/MS). It is known that at least three mechanisms are well accepted for the Biginelli reaction, namely the iminium-, enamine-, and Knoevenagel mechanisms.72 These three mechanisms may be competitive, and the role of catalysis is also to select the reaction pathway in order to improve selectivities.72 ESI-MS(/MS) has been proved to be an outstanding tool for reaction mechanism monitoring73–75 with impressive results for the Biginelli reaction.76,77 We have already used ESI-MS(/MS) as an unsurpassed tool in the mechanistic investigation of the Biginelli reaction.39–41 ESI-MS(/MS) online monitoring was therefore used to investigate the mechanism of the Biginelli reaction using catalyst 1. The model reaction was monitored in the presence of 1 for 30 min, 60 min, 90 min and 120 min (at 100 °C for all times), and the iminium pathway (Scheme 2) was the preferred reaction pathway depicted by ESI-MS(/MS) analyses (Fig. 5).
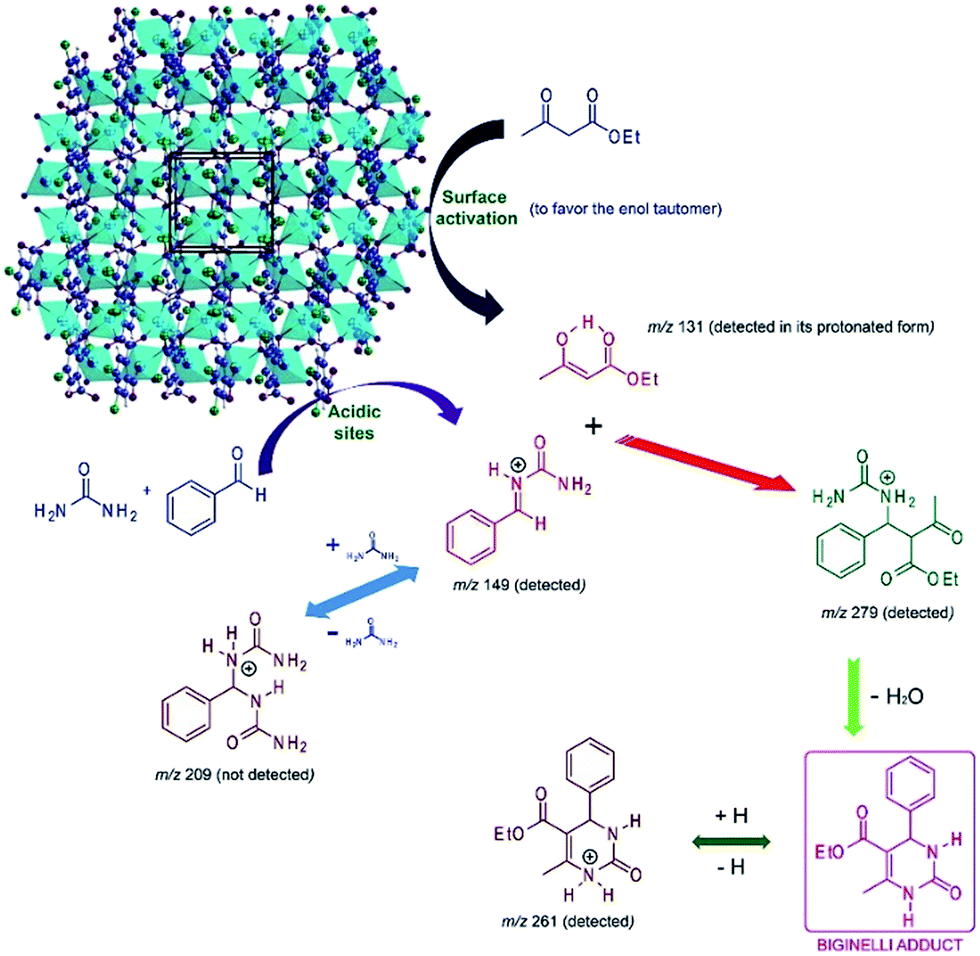 | ||
| Scheme 2 Iminium-like mechanism for the Biginelli reaction catalyzed by CP 1 based on ESI-MS(/MS) analyses. Note the cooperative effect on the formation of the intermediate of m/z 279. Also note that a second urea addition (m/z 209) has not been observed, therefore pointing to the efficiency of the catalytic system towards the Biginelli adduct formation. | ||
 | ||
| Fig. 5 Key intermediates and products detected (and characterized) by ESI(+)-MS(/MS): (I) representative ESI(+)-MS after 90 minutes of online monitoring; (II) ESI(+)-MS/MS of the Knoevenagel intermediate of m/z 219; (III) ESI(+)-MS/MS of key iminium intermediate of m/z 149; (IV) ESI(+)-MS/MS of the intermediate of m/z 173 from the enamine-based mechanism; (V) ESI(+)-MS/MS of the protonated Biginelli adduct of m/z 261. | ||
CP 1 is responsible for activating the 1,3-dicarbonyl derivative (surface activation) affording the more reactive enol tautomer (detected in its protonated form of m/z 131). Catalyst 1 is also responsible for facilitating the iminium ion (m/z 149) key intermediate formation in the acidic sites of CP 1. Once the iminium and the complex are formed, condensation takes place (m/z 279) followed by water release to afford the Biginelli adduct (protonated form of m/z 261). Intermediates from the enamine- (m/z 173) and Knoevenagel (m/z 219) reaction pathways have also been detected, but considering these intermediates are found in equilibrium with the reagents, that during the time monitoring their intensities was very low (hard to acquire their MSMS), and that no further intermediate from these reaction pathways was observed, they may indeed be considered dormant intermediates. The iminium intermediate was the only one detected during the whole reaction time monitoring, with a relatively high intensity at the start of the reaction, thus pointing firmly to its participation in the reaction mechanism. No intermediate from a second urea addition (m/z 209) was observed throughout the monitoring of the Biginelli reaction, indicating therefore the efficiency of the catalyst to further the reaction through the iminium pathway.
The proposed surface activation of the reagents is responsible for the observed change in the structural features of the CP (see Fig. S1†), as depicted from powder X-ray analyses. No metal ion (Zn) could be detected during the ESI-MS(/MS) online monitoring indicating that indeed no metal leaching is taking place. This result is in accordance with the powder X-ray analyses which indicated the phase transition but not the structural collapse of the tested CP in the first catalytic test. Although the primary structure of the catalyst is modified during the reaction, its catalytic activity does not undergo any significant alteration, thus indicating that the catalyst is robust as a promoter of the Biginelli MCR. This observation regarding the structural modification of the hybrid material is overall neglected in the CP/MOF literature. In the current study, the results obtained under flow (or batch) conditions are in accordance with the statement that although the primary structured of the catalyst is modified, its catalytic activity is sustained for at least three runs.
Finally, nine DHPMs had their antitumoral activities evaluated against MCF-7 (human breast cancer cells), A549 (human alveolar basal epithelial cells) and Caco-2 (human epithelial colorectal cells). First, to address the cytotoxicity on normal cells (healthy cells), a primary culture consisting mainly of fibroblasts (healthy cells) was treated with 1.00 mM of the chosen DHPM (4b–j) derivatives for 72 h (Fig. 6).
 | ||
| Fig. 6 Cytotoxicity assays after 72 h of normal cells (fibroblast cells) treatment with DHPM 4b–j. Compounds 4b, 4g and 4h were not cytotoxic for normal cells. The remaining compounds produced cytotoxicity for normal cells at different ranges, but never below 70%. No compound induced more than 40% of cell death. Monastrol (4d) was used as a positive control, and culture medium with diluents (DMSO) was used as negative control. p Value: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Three compounds (4b, 4g and 4h) showed no statistically significant effect on the normal cells, whereas the other six compounds (4c, 4d, 4e, 4f, 4i and 4j) presented different degrees of cytotoxicity in normal cells, but none of them produced cell viability reduction below 70% (see Fig. 6). Additionally, DHPMs 4b–j were tested at high concentrations, so the concentration decrease would be even more beneficial for healthy cells.
The encouraging results prompted us to test the nine DHPMs against tumoral cell models. We started the tests with MCF-7 breast cancer cells. After 24 h and 48 h of treatment, some derivatives produced a statically significant reduction in MCF-7 cell viability (Fig. 7), in accordance with previous results.40 After 72 h of treatment, all compounds caused a statistically significant reduction in cell viability in all tumoral cell samples for MCF-7. For this reason, the DHPM (4b–j) activities were also tested (for 72 h) against A549 and Caco-2 cancer cell lineages (Fig. 7).
 | ||
| Fig. 7 Cytotoxicity assays for MCF-7 (human breast cancer cells), A549 (human alveolar basal epithelial cells) and Caco-2 (human epithelial colorectal cells) at different time periods. p Value: *p < 0.05, **p < 0.01, ***p < 0.001. | ||
Three DHPM derivatives (4c – Oxomonastrol, 4h – Enastron, and 4i – Dimethylenastron) displayed high cytotoxic effects, that is, they induced death in more than 50% of the cells. These derivatives were therefore tested in intermediate concentration from 100 mM to 1 mM in cell models which were susceptible to their effects (A549 and Caco-2 cells) for 72 h of treatment (Fig. 8).
 | ||
| Fig. 8 Cell viability tests with intermediate DHPM concentrations for 72 h of treatment. The three compounds (4c – Oxomonastrol, 4h – Enastron, and 4i – Dimethylenastron) were effective against Caco-2 and A549 cell lineages. p Values: **p < 0.01, ***p < 0.001. | ||
All three compounds showed highly statistically significant cytotoxicity effects against Caco-2 cells in all tested concentrations (Fig. 8). DHPM 4c (Oxomonastrol) was the only one that reduced A549 cell viability (statically significant result) in concentrations above 400 mM (Fig. 8). Compounds 4h (Enastron) and 4i (Dimethylenastron) showed cytotoxic effects in all tested concentrations (Fig. 8). DHPM 4h was also highly effective against all tested cancer cells (Fig. 8) and no cytotoxic effect over normal cells was noted (Fig. 6).
Time-dependent experiments were also conducted and all tested compounds showed activity against A549, Caco-2 and MCF-7 cells (Fig. 9), with a more pronounced effect after 48 h of treatment for A549 and Caco-2 cell models, whereas 72 h of treatment was required to observe similar effects on MCF-7 cells.
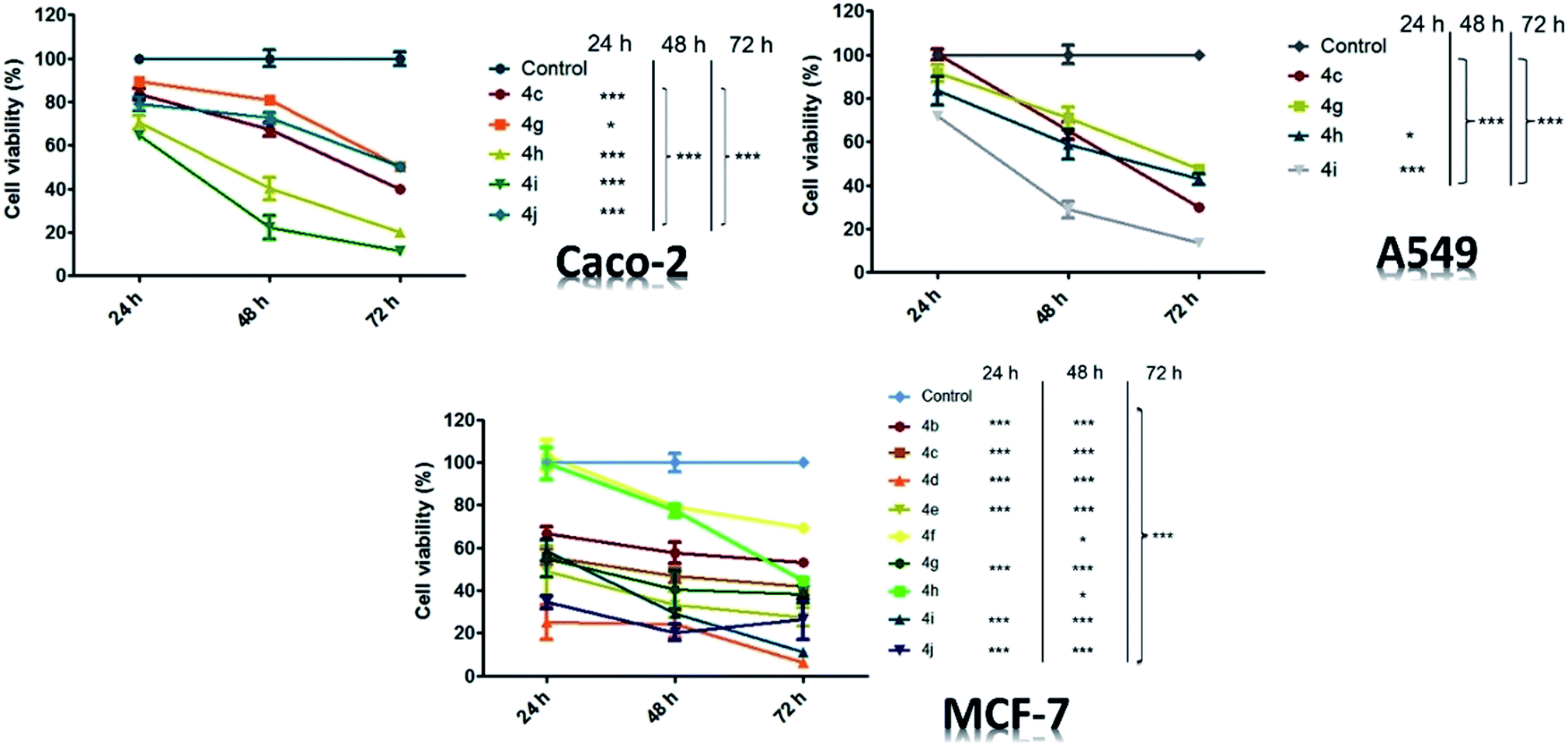 | ||
| Fig. 9 Time-dependent effects for three cell lineages (Caco-2, A549, and MCF-7 cells). p Values: *p < 0.05, ***p < 0.001. | ||
Conclusions
In summary, we have described the synthesis, characterization, photophysical properties and catalytic application of two novel and robust CPs (1 and 2) as heterogeneous catalysts for the Biginelli synthesis under solvent-free conditions and heterogeneous catalysis. The reaction conducted under continuous flow conditions (with DMF) returned very promising results toward a scale-up of the reaction. Furthermore, three bioactive DHPMs have been obtained by this methodology with high yields and short reaction times. The synthesized DHPMs were tested with success with impressive results for Oxomonastrol (4c), Enastron (4h) and Dimethylenastron (4i), which were efficiently used as antitumoral agents against MCF-7, Caco-2 and A549 tumoral cell lineages. In all, the current manuscript provides the baselines for the development of more efficient CPs/MOFs for MCRs, therefore opening up a wide range of opportunities for scale-up processes using continuous flow conditions, and allowing one to envisage new applications for this class of promising materials. In other words, we have described the happy marriage of CPs/MOFs with MCRs.Experimental section
General
Chemicals and solvents were purchased from commercial sources. Liquid reagents and solvents were distilled prior to use.Acknowledgements
The authors gratefully acknowledge CAPES, FAPDF, FAPESP, FINATEC, CNPq (INCT-Inami, INCT-Catalysis and INCT-Transcend group), FACEPE and DPP-UnB for partial financial support.Notes and references
- T. R. Cook, Y.-R. Zheng and P. J. Stang, Chem. Rev., 2012, 113, 734–777 CrossRef PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- S. T. Meek, J. A. Greathouse and M. D. Allendorf, Adv. Mater., 2011, 23, 249–267 CrossRef CAS PubMed.
- J. Rocha, L. D. Carlos, F. A. A. Paz and D. Ananias, Chem. Soc. Rev., 2011, 40, 926–940 RSC.
- J. A. Mason, M. Veenstra and J. R. Long, Chem. Sci., 2014, 5, 32–51 RSC.
- N. A. Khan, Z. Hasan and S. H. Jhung, J. Hazard. Mater., 2013, 244, 444–456 CrossRef PubMed.
- J. Gascon, A. Corma, F. Kapteijn and F. X. Llabrés i Xamena, ACS Catal., 2014, 4, 361–378 CrossRef CAS.
- A. Dhakshinamoorthy, M. Alvaro and H. Garcia, Chem. Commun., 2012, 48, 11275–11288 RSC.
- L. Q. Ma, C. Abney and W. B. Lin, Chem. Soc. Rev., 2009, 38, 1248–1256 RSC.
- F. Zaera, ChemSusChem, 2013, 6, 1797–1820 CrossRef CAS PubMed.
- F. A. A. Paz, J. Klinowski, S. M. F. Vilela, J. P. C. Tome, J. A. S. Cavaleiro and J. Rocha, Chem. Soc. Rev., 2012, 41, 1088–1110 RSC.
- R. J. Kuppler, D. J. Timmons, Q. R. Fang, J. R. Li, T. A. Makal, M. D. Young, D. Q. Yuan, D. Zhao, W. J. Zhuang and H. C. Zhou, Coord. Chem. Rev., 2009, 253, 3042–3066 CrossRef CAS PubMed.
- M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151–1152 CrossRef CAS.
- A. Dhakshinamoorthy and H. Garcia, Chem. Soc. Rev., 2014, 43, 5750–5765 RSC.
- Z.-J. Lin, J. Lu, M. Hong and R. Cao, Chem. Soc. Rev., 2014, 43, 5867–5895 RSC.
- J. Liu, L. Chen, H. Cui, J. Zhang, L. Zhang and C.-Y. Su, Chem. Soc. Rev., 2014, 43, 6011–6061 RSC.
- Q. Zhang, E. Uchaker, S. L. Candelaria and G. Cao, Chem. Soc. Rev., 2013, 42, 3127–3171 RSC.
- A. Domling, W. Wang and K. Wang, Chem. Rev., 2012, 112, 3083–3135 CrossRef CAS PubMed.
- S. Brauch, S. S. van Berkel and B. Westermann, Chem. Soc. Rev., 2013, 42, 4948–4962 RSC.
- D. Bonne, T. Constantieux, Y. Coquerel and J. Rodriguez, Chem.–Eur. J., 2013, 19, 2218–2231 CrossRef CAS PubMed.
- X. Guo and W. H. Hu, Acc. Chem. Res., 2013, 46, 2427–2440 CrossRef CAS PubMed.
- J. Wegner, S. Ceylan and A. Kirschning, Adv. Synth. Catal., 2012, 354, 17–57 CrossRef CAS PubMed.
- A. K. Gupta, N. Singh and K. N. Singh, Curr. Org. Chem., 2013, 17, 474–490 CrossRef CAS.
- G. C. Tron, A. Minassi and G. Appendino, Eur. J. Org. Chem., 2011, 5541–5550 CrossRef CAS PubMed.
- C. O. Kappe, Eur. J. Med. Chem., 2000, 35, 1043–1052 CrossRef CAS.
- J. E. Biggs-Houck, A. Younai and J. T. Shaw, Curr. Opin. Chem. Biol., 2010, 14, 371–382 CrossRef CAS PubMed.
- P. Slobbe, E. Ruijter and R. V. A. Orru, Med. Chem. Commun., 2012, 3, 1189–1218 RSC.
- A. Crespo, A. El Maatougui, P. Biagini, J. Azuaje, A. Coelho, J. Brea, M. I. Loza, M. I. Cadavid, X. García-Mera, H. Gutiérrez-de-Terán and E. Sotelo, ACS Med. Chem. Lett., 2013, 4, 1031–1036 CrossRef CAS PubMed.
- T. Yang, H. Cui, C. Zhang, L. Zhang and C.-Y. Su, ChemCatChem, 2013, 5, 3131–3138 CrossRef CAS PubMed.
- S. Rostamnia, H. Xin and N. Nouruzi, Microporous Mesoporous Mater., 2013, 179, 99–103 CrossRef CAS PubMed.
- M. J. Climent, A. Corma and S. Iborra, RSC Adv., 2012, 2, 16–58 RSC.
- P. Li, S. Regati, R. J. Butcher, H. D. Arman, Z. X. Chen, S. C. Xiang, B. L. Chen and C. G. Zhao, Tetrahedron Lett., 2011, 52, 6220–6222 CrossRef CAS PubMed.
- C. M. MacNeill, C. S. Day, A. Marts, A. Lachgar and R. E. Noftle, Inorg. Chim. Acta, 2011, 365, 196–203 CrossRef CAS PubMed.
- S. Rostamnia and A. Morsali, RSC Adv., 2014, 4, 10514–10518 RSC.
- M. De Toni, R. Jonchiere, P. Pullumbi, F.-X. Coudert and A. H. Fuchs, ChemPhysChem, 2012, 13, 3497–3503 CrossRef CAS PubMed.
- S. S. Han, S.-H. Choi and A. C. T. van Duin, Chem. Commun., 2010, 46, 5713–5715 RSC.
- S. S. Kaye, A. Dailly, O. M. Yaghi and J. R. Long, J. Am. Chem. Soc., 2007, 129, 14176–14177 CrossRef CAS PubMed.
- G. A. Medeiros, W. A. da Silva, G. A. Bataglion, D. A. C. Ferreira, H. C. B. de Oliveira, M. N. Eberlin and B. A. D. Neto, Chem. Commun., 2014, 50, 338–340 RSC.
- H. G. O. Alvim, T. B. de Lima, H. C. B. de Oliveira, F. C. Gozzo, J. L. de Macedo, P. V. Abdelnur, W. A. Silva and B. A. D. Neto, ACS Catal., 2013, 3, 1420–1430 CrossRef CAS.
- L. M. Ramos, B. C. Guido, C. C. Nobrega, J. R. Corrêa, R. G. Silva, H. C. B. de Oliveira, A. F. Gomes, F. C. Gozzo and B. A. D. Neto, Chem.–Eur. J., 2013, 19, 4156–4168 CrossRef CAS PubMed.
- L. M. Ramos, A. Tobio, M. R. dos Santos, H. C. B. de Oliveira, A. F. Gomes, F. C. Gozzo, A. L. de Oliveira and B. A. D. Neto, J. Org. Chem., 2012, 77, 10184–10193 CrossRef CAS PubMed.
- I. T. Weber, I. A. A. Terra, A. J. G. d. Melo, M. A. d. M. Lucena, K. A. Wanderley, C. d. O. Paiva-Santos, S. G. Antonio, L. A. O. Nunes, F. A. A. Paz, G. F. d. Sa, S. A. Junior and M. O. Rodrigues, RSC Adv., 2012, 2, 3083–3087 RSC.
- I. B. Vasconcelos, T. G. da Silva, G. C. G. Militao, T. A. Soares, N. M. Rodrigues, M. O. Rodrigues, N. B. da Costa Jr, R. O. Freire and S. A. Junior, RSC Adv., 2012, 2, 9437–9442 RSC.
- I. T. Weber, A. J. G. de Melo, M. A. de Melo Lucena, M. O. Rodrigues and S. Alves Junior, Anal. Chem., 2011, 83, 4720–4723 CrossRef CAS PubMed.
- X.-H. Lou, C. Xu, H.-M. Li, Z.-Q. Wang, H. Guo and D.-X. Xue, CrystEngComm, 2013, 15, 4606–4610 RSC.
- Y. Y. Xu, X. X. Wu, Y. Y. Wang, X. M. Su, S. X. Liu, Z. Z. Zhu, B. Ding, Y. Wang, J. Z. Huo and G. X. Du, RSC Adv., 2014, 4, 25172–25182 RSC.
- J. Xia, Z.-j. Zhang, W. Shi, J.-f. Wei and P. Cheng, Cryst. Growth Des., 2010, 10, 2323–2330 CAS.
- W. W. Lestari, H. C. Streit, P. Lonnecke, C. Wickleder and E. Hey-Hawkins, Dalton Trans., 2014, 8188–8195 RSC.
- W. W. Lestari, P. Lonnecke, M. B. Sarosi, H. C. Streit, M. Adlung, C. Wickleder, M. Handke, W.-D. Einicke, R. Glaser and E. Hey-Hawkins, CrystEngComm, 2013, 15, 3874–3884 RSC.
- C. O. Kappe, Tetrahedron, 1993, 49, 6937–6963 CrossRef CAS.
- P. Lacotte, D. A. Buisson and Y. Ambroise, Eur. J. Med. Chem., 2013, 62, 722–727 CrossRef CAS PubMed.
- R. F. S. Canto, A. Bernardi, A. M. O. Battastini, D. Russowsky and V. L. Eifler-Lima, J. Braz. Chem. Soc., 2011, 22, 1379–1388 CrossRef CAS PubMed.
- H. Y. K. Kaan, V. Ulaganathan, O. Rath, H. Prokopcova, D. Dallinger, C. O. Kappe and F. Kozielski, J. Med. Chem., 2010, 53, 5676–5683 CrossRef CAS PubMed.
- M. Pramanik and A. Bhaumik, ACS Appl. Mater. Interfaces, 2014, 6, 933–941 CAS.
- X.-L. Shi, X. Xing, H. Lin and W. Zhang, Adv. Synth. Catal., 2014, 356, 2349–2354 CrossRef CAS PubMed.
- A. Mobaraki, B. Movassagh and B. Karimi, Appl. Catal., A, 2014, 472, 123–133 CrossRef CAS PubMed.
- V. Escande, L. Garoux, C. Grison, Y. Thillier, F. Debart, J.-J. Vasseur, C. Boulanger and C. Grison, Appl. Catal., B, 2014, 146, 279–288 CrossRef CAS PubMed.
- A. Q. Wang, X. Liu, Z. X. Su and H. W. Jing, Catal. Sci. Technol., 2014, 4, 71–80 CAS.
- D. Elhamifar and A. Shábani, Chem.–Eur. J., 2014, 20, 3212–3217 CrossRef CAS PubMed.
- B. Karimi, A. Mobaraki, H. M. Mirzaei, D. Zareyee and H. Vali, ChemCatChem, 2014, 6, 212–219 CrossRef CAS PubMed.
- J. Safari and S. Gandomi-Ravandi, RSC Adv., 2014, 4, 11486–11492 RSC.
- Z. N. Siddiqui and T. Khan, RSC Adv., 2014, 4, 2526–2537 RSC.
- J. Safari and Z. Zarnegar, New J. Chem., 2014, 38, 358–365 RSC.
- M. Oliverio, P. Costanzo, M. Nardi, I. Rivalta and A. Procopio, ACS Sustainable Chem. Eng., 2014, 2, 1228–1233 CrossRef CAS.
- Y. L. Gu, Green Chem., 2012, 14, 2091–2128 RSC.
- M. S. Singh and S. Chowdhury, RSC Adv., 2012, 2, 4547–4592 RSC.
- K. Shanab, C. Neudorfer, E. Schirmer and H. Spreitzer, Curr. Org. Chem., 2013, 17, 1179–1187 CrossRef CAS.
- M. B. Gawande, V. D. B. Bonifácio, R. Luque, P. S. Branco and R. S. Varma, ChemSusChem, 2014, 7, 24–44 CrossRef CAS PubMed.
- C. Wiles and P. Watts, Green Chem., 2014, 16, 55–62 RSC.
- T. N. Glasnov, D. J. Vugts, M. M. Koningstein, B. Desai, W. M. F. Fabian, R. V. A. Orru and C. O. Kappe, QSAR Comb. Sci., 2006, 25, 509–518 CAS.
- T. N. Glasnov and C. O. Kappe, J. Heterocycl. Chem., 2011, 48, 11–30 CrossRef CAS PubMed.
- H. G. O. Alvim, T. B. Lima, A. L. de Oliveira, H. C. B. de Oliveira, F. M. Silva, F. C. Gozzo, R. Y. Souza, W. A. da Silva and B. A. D. Neto, J. Org. Chem., 2014, 79, 3383–3397 CrossRef CAS PubMed.
- F. Coelho and M. N. Eberlin, Angew. Chem., Int. Ed., 2011, 50, 5261–5263 CrossRef CAS PubMed.
- L. S. Santos, J. Braz. Chem. Soc., 2011, 22, 1827–1840 CrossRef CAS PubMed.
- L. S. Santos, Eur. J. Org. Chem., 2008, 235–253 CrossRef CAS PubMed.
- R. De Souza, E. T. da Penha, H. M. S. Milagre, S. J. Garden, P. M. Esteves, M. N. Eberlin and O. A. C. Antunes, Chem.–Eur. J., 2009, 15, 9799–9804 CrossRef CAS PubMed.
- M. K. Raj, H. S. P. Rao, S. G. Manjunatha, R. Sridharan, S. Nambiar, J. Keshwan, J. Rappai, S. Bhagat, B. S. Shwetha, D. Hegde and U. Santhosh, Tetrahedron Lett., 2011, 52, 3605–3609 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures and spectroscopic data for all compounds. CCDC 1020344 and 1020345. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra07677c |
| This journal is © The Royal Society of Chemistry 2015 |