DOI:
10.1039/C5RA05841D
(Paper)
RSC Adv., 2015,
5, 48500-48505
Superhydrophobic surface formation and modulation in a biphenyltetracarboxylic dianhydride derivative self-assembly system via changing alkyl chain lengths†
Received
2nd April 2015
, Accepted 21st May 2015
First published on 21st May 2015
Abstract
A series of organogelators (C4, C6, C12 and C18) based on biphenyltetracarboxylic dianhydride derivative were designed and synthesized. The organogels could be obtained via self-assembly of the biphenyltetracarboxylic dianhydride derivatives in some frequently-used solvents. The organogels were thoroughly characterized using various microscopic techniques including field-emission scanning electron microscopy (FESEM), X-ray diffraction (XRD), UV-vis and fluorescence spectra, contact angle. Interestingly, superhydrophobic surface was formed via the self-assembly of compound C12 in petroleum ether and exhibited the lotus-effect. The surface wettability could be modulated via changing alkyl chain lengths. The π–π stacking and van der Waals force were possible the main driving forces for gel formation. This gel system held promise for soft materials application in upscale superhydrophobic surface materials.
Introduction
The surface structure of the lotus leaf exhibits superhydrophobicity and the self-cleaning property. Inspired by ‘‘the lotus effect’’ in nature, superhydrophobic surfaces (a water contact angle >150° and a low roll-off angle) have gained much attention due to their potential applications in anti-corrosive coating, anti icing, frictional force reduction, self-cleaning, and fluidic channels.1 At the same time, superhydrophobic surfaces also can be endowed with more and more functionalities such as low-/high-adhesion, anisotropy, antibacterial, antiplatelet, chirality, drug reduction, water-collecting, electro-wetting, anti-reflection, structural color, and transparency.2–4 So, many artificial hydrophobic surfaces are fabricated by inorganic, composite materials and polymer materials such as ZnO, SiO2, CuO, TiO2 and so on.5–9 Comparing with the inorganic materials, organic materials provide much more modification chances for preparation of superhydrophobic surfaces. Especially, supramolecular chemistry provides an organized molecular architecture which gives a good paradigm for biomimicry. Supramolecular gels formed by specific non-covalent interactions including hydrogen bonds, hydrophobic interactions, pie–pie interactions, and van der Waals forces have been extensively studied in the past decades.10–15 Supramolecular gel was a class of soft matter. The advantages of gel were the bionic function and plastically. A various of exciting nanostructures ranging from nanofibre, nanowhisker, nanotube, nanoball, liquid crystal, vesicle and crystalline networks are prepared in the supramolecular self-assembly process.16 The different nanostructures can be possible exhibited superhydrophobic properties. Yi and co-workers reported that xerogels formed from cholesterol-based organogels could also express hydrophobic surfaces.17–19 Nakano and coworker prepared superhydrophobic surfaces by fibrous aggregation of perfluoroalkyl chain-containing organogelators.20 The water contact angles of superhydrophobic surfaces were greater than 150°. A new C60 compound with a L-glutamid-derived lipid unit was self-assembled into organogel.21 The xerogel film of the low molecule weight gel from toluene showed a water contact angle of 142.0°.
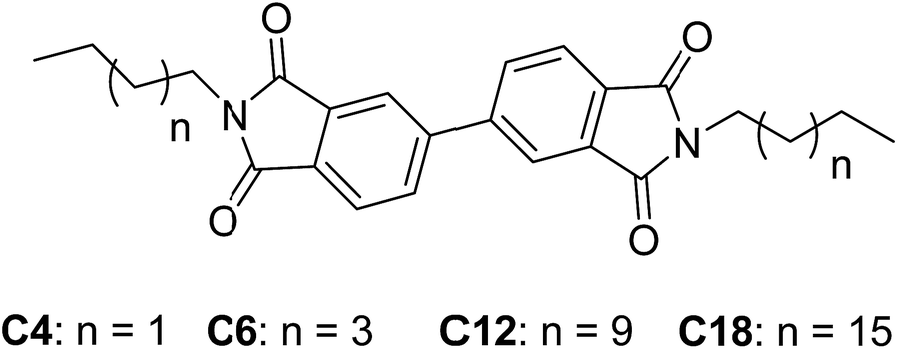
In this paper, a series of new 3,3′,4,4′-biphenyltetracarboxylic -based organogelators C4, C6, C12 and C18 with the different alkyl chain were synthesized and studied (Scheme 1). The organogelators can be self-assembled into organogel in some frequently-used solvents. The different nanostructures were obtained in the self-assembly process. At the same time, the xerogel film from petroleum ether exhibited superhydrophobic. So the organogels of C6, C12 and C18 from petroleum ether were studied in detail.
 |
| | Scheme 1 The chemical structure of organogels C4, C6, C12 and C18. | |
Experimental procedure
Reagents and solutions
n-Butylamine, hexylamine, dodecylamine, octadecylamine and 3,3′,4,4′-biphenyltetracarboxylic dianhydride (98%) were supplied from Zhengzhou Alfachem Co., Ltd. Organic solvents, i.e. acetonitrile, ethanol, Et3N and CH2Cl2 were obtained from Sinopharm Chemical Reagent Co. Ltd. All other reagents were of analytical grade.
Synthesis of organogelator
Compound C4, C6, C12 and C18 were prepared using the 3,3′,4,4′-biphenyltetracarboxylic dianhydride reaction with organic amine with the corresponding carbon chain in ethanol. The details of the synthesis are as followings.
Synthesis of C4. 3,3′,4,4′-biphenyltetracarboxylic dianhydride (2.1 g, 7.1 mmol) and hexylamine (1.14 g, 15.7 mmol) were added to 80 mL ethanol. The mixture was stirred and heated to reflux. When the solid was completely dissolved, then the solution was cooled to room temperature and large amount solid was appeared in the above solution. The above mixture was filtered and washed by ethanol for three times. The white solid was the compound C4 with the yield of 81%; 1H NMR (400 MHz, CDCl3): δ 8.093 (s, 2H), 7.965 (broad s, 4H), 3.715 (t, 4H, J = 7.6 Hz), 1.695 (m, 4H), 1.327 (m, 4H), 0.886 (t, 6H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3): δ 168.0, 140.1, 132.7, 133.1, 131.5, 131.2, 131.0, 128.3, 130.2, 128.2, 128.1, 40.0, 33.9, 27.5, 22.7, 14.0; ESI-MS (m/z): [M + H]+ calcd for C24H25N2O4: 405.1814. Found: 405.1810.
Synthesis of C6. 3, 3′, 4, 4′-biphenyltetracarboxylic dianhydride (2.0 g, 6.8 mmol) and hexylamine (1.51 g, 14.9 mmol) were added to 100 mL ethanol. The mixture was stirred and heated to reflux. When the solid was completely dissolved, then the solution was cooled to room temperature and large amount solid was appeared in the above solution. The above mixture was filtered and washed by ethanol for three times. The white solid was the compound C6 with the yield of 75%; 1H NMR (400 MHz, CDCl3): δ 8.093 (s, 2H), 7.965 (broad s, 4H), 3.716 (t, 4H, J = 7.6 Hz), 1.696 (m, 4H), 1.327 (m, 12H), 0.886 (t, 6H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3): δ 167.9, 140.0, 132.6, 133.2, 131.4, 131.2, 131.0, 128.4, 130.2, 128.1, 128.0, 40.1, 33.9, 31.6, 27.4, 26.5, 22.8, 14.1; ESI-MS (m/z): [M + H]+ calcd for C28H33N2O4: 461.2440. Found: 461.2429.
Synthesis of C12. 3,3′,4,4′-biphenyltetracarboxylic dianhydride (1.0 g, 3.4 mmol) and dodecylamine (1.39 g, 7.5 mmol) were added to 100 mL ethanol. The mixture was stirred and heated to reflux. When the solid was completely dissolved, then the solution was cooled to room temperature and large amount solid was appeared in the above solution. The above mixture was filtered and washed by ethanol for three times. The white solid was the compound C12 with the yield of 70%; 1H NMR (400 MHz, CDCl3): δ 8.092 (s, 2H), 7.964 (broad s, 4H), 3.713 (t, 4H, J = 7.6 Hz), 1.693 (m, 4H), 1.337 (m, 4H), 1.249 (m, 32H), 0.873 (t, 6H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3): δ 167.8, 140.1, 132.5, 133.1, 131.3, 131.1, 131.1, 128.3, 130.1, 128.1, 128.0, 40.2, 31.9, 29.7, 29.4, 27.4, 26.8, 24.2, 22.7, 14.0; ESI-MS (m/z): [M + H]+ calcd for C40H57N2O4: 629.4318; found: 629.4325.
Synthesis of C18. 3,3′,4,4′-biphenyltetracarboxylic dianhydride (1.0 g, 3.4 mmol) and octadecylamine (2.17 g, 7.5 mmol) were added to 100 mL ethanol. The mixture was stirred and heated to reflux. When the solid was completely dissolved, then the solution was cooled to room temperature and large amount solid was appeared in the above solution. The above mixture was filtered and washed by ethanol for three times. The white solid was the compound C18 with the yield of 72%; 1H NMR (400 MHz, CDCl3): δ 8.094 (s, 2H), 7.964 (broad s, 4H), 3.714 (t, 4H, J = 7.6 Hz), 1.694 (m, 4H), 1.249 (m, 60H), 0.876 (t, 6H, J = 6.8 Hz); 13C NMR (100 MHz, CDCl3): δ 167.8, 140.1, 132.7, 133.3, 131.5, 131.3, 131.1, 128.5, 130.3, 128.2, 128.1, 40.2, 31.8, 29.8, 29.5, 27.5, 26.9, 22.8, 14.1; ESI-MS (m/z): [M + H]+ calcd for C52H81N2O4: 797.6196; found: 797.6197.
The gelation test
The gelation test on C4, C6, C12 and C18 (25 mg mL−1) was carried out with various solvents using a test tube inversion method.17 The tube was heated to 80 °C, and then put into a thermostat controlled by water (25 °C). Qualitatively, gelation was considered successful if no sample flow was observed upon inversion of the container at room temperature (the inverse flow method).
Techniques
The 1H NMR (400 MHz) and 13C NMR (100 MHz) spectra were recorded on a Mercury plus-Varian instrument. Proton chemical shifts are reported in parts per million downfield from tetramethylsilane (TMS). HRMS was recorded on LTQ-Orbitrap mass spectrometer (ThermoFIsher, San Jose, CA). SEM images were obtained using a FE-SEM S-4800 (Hitachi) instrument. Samples were prepared by spinning the diluted gels on glass slices and coating with Au. Powder X-ray diffractions were generated by using a Philips PW3830 sealed-tube X-ray generator (Cu target, λ = 0.1542 nm) with a power of 40 kV and 50 mA. UV-vis absorption was recorded on a UV-vis 2550 spectroscope (Shimadzu). Fluorescent spectra were recorded on Edinburgh Instruments FLS 900 (Edinburgh Instruments, Ltd. Livingston, UK). Water contact angles were performed using the sessile drop method (Dataphysics, OCA 20). The water droplets were introduced using a microsyringe, and images were captured to measure the angle of the liquid solid interface; each sample was recorded at three different points.
Results and discussion
The gelation ability of organogelators
The gelation ability of gelators C4, C6, C12 and C18 was tested in fourteen kinds of frequently-used organic solvent. The experiment results were listed in Table 1. The gelation ability of the four gelators was different because of their different length of alkyl chain. The gelator C4 could not effectively form organogel in the experimental solvents. The partial gels were formed in some solvents such as acetonitrile, acetone, toluene, ethyl acetate, ethanol and DMSO. The gelator C6 could form organogel only in three kinds of solvent, such as acetone, petroleum ether and DMSO with the critical gelation concentration of 12.5, 25 and 25 mg mL−1, respectively. With the length of alkyl chain increasing, the gelator C12 could form organogel in five kinds of solvent, such as methanol, ethanol, ethyl acetate, petroleum ether and DMSO with the critical gelation concentration of 25, 25, 25, 25 and 12.5 mg mL−1, respectively. When the length of alkyl chain was increased to 18, the gelator C18 could gelate six kinds of solvent such as acetonitrile, 1, 4-dioxane, n-hexane, ethyl acetate, ethanol and petroleum ether with the critical gelation concentration of 25, 25, 5, 25, 25 and 6.25 mg mL−1, respectively. The critical gelation concentration (CGC) of the gelator in corresponding solvents was listed in brackets of the table. The length of alkyl chain produced some effect on the gelation ability based on the above results. In order to compare and study the oganogel properties, the solvent of petroleum ether was selected and shared for 3,3′,4,4′-biphenyltetracarboxylic dianhydride derivatives gel system. The images of the organogels C6, C12 and C18 in petroleum ether were showed in Fig. 1. The gelators C6, C12 and C18 all formed opaque organogel in petroleum ether. The organogels C6, C12 and C18 could emit blue light under the irradiation of 365 nm light.
Table 1 Gelation properties of compound C6, C12 and C18 in various solventsa
| Solvent |
C4 |
C6 |
C12 |
C18 |
| NI = not soluble; S = solution; PG = partial gel; G = gel; the values in the brackets denote the CGC. |
| Methanol |
NI |
PG |
G (25) |
PG |
| DMF |
S |
S |
PG |
PG |
| Acetonitrile |
PG |
PG |
PG |
G (25) |
| 1,4-Dioxane |
S |
PG |
PG |
G (25) |
| n-hexane |
NI |
PG |
PG |
G (5) |
| Acetone |
PG |
G (12.5) |
PG |
PG |
| Toluene |
PG |
PG |
PG |
PG |
| Ethyl acetate |
PG |
PG |
G (25) |
G (25) |
| Ethanol |
PG |
PG |
G (25) |
G (25) |
| Petroleum ether |
NI |
G (25) |
G (25) |
G (6.25) |
| DMSO |
PG |
G (25) |
G (12.5) |
PG |
| THF |
S |
S |
S |
S |
| CH2Cl2 |
S |
S |
S |
S |
| CHCl3 |
S |
S |
S |
S |
 |
| | Fig. 1 Images of gel C6, C12 and C18 in petroleum ether with the concentration of 25, 25 and 6.25 mg mL−1 (a) the gel C6; (a′) the gel C6 under the irradiation of 365 nm light; (b) the gel C12; (b′) the gel C12 under the irradiation of 365 nm light; (c) the gel C18; (c′) the gel C18 under the irradiation of 365 nm light. | |
Morphology of the self-assembly
Various morphologies could be obtained in the self-assembly process of low molecule weight compound via intermolecular noncovalent interaction. The organogels C6, C12 and C18 in different solvent were observed by field emission scanning electron microscope (FESEM). The fibre structure was obtained in the organogel C6 from petroleum ether (Fig. 2a and a′). The fibre with the diameter of about 100 nm and the length of several micrometers was neatly arranged. The fibre structure was also observed in the organogel C12 from petroleum ether (Fig. 2b and b′). It was different that the length and width of the fibre was smaller than that of the organogel C6. The length and diameter of the fibre in the organogel C12 were about 2 μm and 60 nm, respectively. The fibre from organogel C6 was obvious longer than that of organogel C12. At the same time, the roughness in organogel C12 was larger than that of organogel C6. A distinct structure was obtained in the organogel C18 from petroleum ether. An irregular flaky texture with a width or length of around several hundred nm to several micrometers was revealed in the organogel C18 from petroleum ether (Fig. 2c and c′). The flaky texture structure in organogel C18 gave a little roughness. The roughness in materials surface could produce significant effect on the surface wettability. The morphology of oraganogels C6, C12 and C18 from other solvents was investigated via FESEM and showed in Fig. S1–S3.† Organogel C6 in acetone and DMSO showed microbelts and sheet structure, respectively in Fig. S1.† Belt or sheet structures were also revealed in organogel C12 from the other solvents (Fig. S2†). The irregular flaky texture was still existed in organogel C18 in other solvents (Fig. S3†).
 |
| | Fig. 2 SEM images of the xerogels in petroleum ether at room temperature (25 °C); (a) and (a′) for C6; (b) and (b′) for C12; (c) and (c′) for C18. The concentrations of the gels are 25, 25 and 6.25 mg mL−1 of 1, scale bar for a, a′, b, b′, c and c′ are 5.0, 1.0, 5.0, 2.0, 5.0 and 2.0 μm, respectively. | |
Absorption and emission spectra
In order to under the finer details self-assembly process, the UV-vis spectra of the solution and gel states was carried out and showed in Fig. 3 and S4a.† The compound C4, C6, C12 and C18 in the petroleum ether solution with the concentration of 10−5 mol L−1 all exhibited three absorption bands at 234, 254 and 316 nm. The absorption bands at 234 and 254 nm were related to π–π* transition bands.22,23 The absorption band of 316 nm was related to the n–π* transition band.24,25 The absorption spectra were all broadened and red-shifted comparing with the solution state. The absorption bands of the gel state were red-shifted and broadened to 277 and 353 nm for compound C6. The absorption bands of the gel state were red-shifted and broadened to 249 and 328 nm for compound C12. The absorption bands of the gel state were red-shifted and broadened to 251 and 331 nm for compound C18. It is well known that self-assembly of the molecules by a pi-stacking process may be classified as J or H aggregates. In the former, side-by-side stacking occurs while in the latter, face-to-face stacking occurs.26–29 On the basis of the above experimental results, the UV-vis absorption spectra of organogel C6, C12 and C18 indicated that the π–π stacking was occurred between two neighbouring molecules. At the same time, the J-type aggregation mode was employed in the self-assembly process.
 |
| | Fig. 3 The absorption spectra for compound C6, C12 and C18 in petroleum ether solution and gel states: (a) for C6; (b) for C12; (c) for C18; The solution concentration of compound C6, C12 and C18 was all 1.0 × 10−5 mol L−1; the concentration of the organogel C6, C12 and C18 was 25, 25 and 6.25 mg mL−1, respectively. The UV-vis absorption intensity was normalized. | |
The fluorescent spectra of C6, C12 and C18 gave more information about the aggregation mode in gel C6, C12 and C18 (Fig. 4). The fluorescence emission spectra of compound C6, C12 and C18 in the solution of petroleum ether with the concentration of 10−5 mol L−1 were at 327 nm. The fluorescence emission of compound C4 in solution state was also investigated and showed in Fig. S4b.† The compound C4 in solution with the concentration of 10−5 mol L−1 also had a emission band at 327 nm. When the organogels C6 and C12 were formed in petroleum ether with the concentration of 25 mg mL−1, the fluorescence emission was red-shifted to 392 and 395 nm, respectively. Interestingly, the fluorescence emission of organogel C18 with a low concentration of 6.25 mg mL−1 had a larger red-shifting of 87 nm than the other organogels. These results further indicated that π–π stacking was the main driving force for the gel formation. When the length of alkyl chain was increased, the fluorescence emission had an obvious red-shift comparing with the compound with short alkyl chain in the same state.30 So the longer alkyl chain in this style compound structure was possible to promote the pi–pi stacking to some extent.
 |
| | Fig. 4 The emission spectra for compound C6, C12 and C18 in petroleum ether solution and gel states (λex = 300 nm): (a) for C6; (b) for C12; (c) for C18; the solution concentration of compound C6, C12 and C18 was all 1.0 × 10−5 mol L−1; the concentration of the organogel C6, C12 and C18 was 25, 25 and 6.25 mg mL−1, respectively. The fluorescence emission intensity was normalized. | |
Wettability of the surfaces
Superhydrophobic surfaces have come to the forefront of research due to their great potential application in daily life, industry and agriculture.31,32 These materials have well-defined micro- and nanostructures on their surfaces. The wetting properties of the surfaces from the xerogel film were examined by means of the contact angle measurements. when a xerogel of C6 from petroleum ether was directly coated onto a glass slice, the film gave a hydrophobic surface with a contact angle of 143.1°, advancing contact angle of 161.5°, and receding contact angle of 143.8° (Fig. 5, C6, C6-a and C6-r). The contact angle hysteresis, H (the difference between the advancing contact angle and the receding contact angle), was 17.7°. A superhydrophobic surface with a contact angle of 154.3° was obtained in the organogel C12 from petroleum ether. The advancing and receding contact angles were 156.7° and 147.1°, and the contact angle hysteresis was 9.6°. The water drop on the film of xerogel C12 could freely roll like water on lotus leaf when the sheet glass was pushed with hand, and the experiment process was shot in a movie in the ESI.† The morphology of organogel C6 and C12 was so similar that their wettability properties of the surfaces of the xerogel were almost the same. At the same time, the superhydrophobic surface behaviour for xerogel C12 over time was observed and showed in Fig. S5.† The fresh simple from C12 was place in the air for a month. The sample surface wetability was tested after a week and a month later. The contact angle was 153.6° and 152.1°, respectively. This result showed the superhydrophobic surface from xerogel C12 had good stability. The morphology of organogel C18 was obviously different from that of organogel C6 and C12. So the contact angle of surface from xerogel C18 was 123.4° which was obvious smaller than that of the other two organogelators in Fig. S6.† The advancing and receding contact angles were not obtained because the sliding angle was more than 90°. The materials surface wettability was determined by the chemical composition and microstructure on the surface. For this self-assembly system, the chemical composition was very similar. Then the difference of the microstructure of different samples was the reason for the different hydrophobicity. The hydrophobicity of xerogel film from C12 was superior to that of xerogel film from C6. It was possible due to the length and width of the fibre from organogel C12 was smaller than that of the organogel C6. So, the stacking of the fibre played the role as the lotus leaf surface mastoid.33 When an irregular flaky texture was formed in the organogel C18, the hydrophobicity was naturally not well. The superhydrophobic surface can be prepared via a simple method of supramolecular self-assembly.
 |
| | Fig. 5 Water contact angle experiments results of the film coating with xerogel C6 and C12 from petroleum ether: C6 and C12 for contact angle; C6-a and C12-a for advancing contact angle; C6-r and C12-r for receding contact angle. | |
X-ray diffraction (XRD) study and DFT calculations
To gain a deeper insight into the self-assembly mechanism, small angle X-ray scattering (SAXS) and large powder X-ray diffraction experiments were also carried out (Fig. S7a and b†). The SAXS experiment of organogel C18 exhibited two peaks with the d-space values of 53.8 and 33.3 Å.34–36 There were no peaks in the SAXS experiment of organogel C6 and C12. The large powder X-ray diffraction experiment results were showed in Fig. S6b.† The existence of a series of peaks corresponding to distances of 14.4, 8.1 and 5.8 Å with the ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: :
: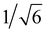 in organogel C6, indicated a more complicated structure (Fig. S7b†). There were few peaks for organogel C12 and C18. Insight into the molecule structure in gel state, the optimized conformation of the truncated models (alkyl chains are replaced by Me groups) of those compounds in monomer was gained using the hybrid B3LYP density functional theory method with the 6-31+G(d) basis set.37,38 Optimized structures (B3LYP/6-31G+(d)) was showed in Fig. 6. The dihedral angle of the two benzene rings of the molecule C6, C12 and C18 was 39.3°. This result well explained the difference of fluorescence emission of organogels C6, C12 and C18 in petroleum ether. If the two benzene rings were in a plane, then the fluorescence emission of organogels C6 and C12 would be much the same as that of organogel C18. The length of alkyl chain had not obvious effect on the organogel fluorescence emission.
in organogel C6, indicated a more complicated structure (Fig. S7b†). There were few peaks for organogel C12 and C18. Insight into the molecule structure in gel state, the optimized conformation of the truncated models (alkyl chains are replaced by Me groups) of those compounds in monomer was gained using the hybrid B3LYP density functional theory method with the 6-31+G(d) basis set.37,38 Optimized structures (B3LYP/6-31G+(d)) was showed in Fig. 6. The dihedral angle of the two benzene rings of the molecule C6, C12 and C18 was 39.3°. This result well explained the difference of fluorescence emission of organogels C6, C12 and C18 in petroleum ether. If the two benzene rings were in a plane, then the fluorescence emission of organogels C6 and C12 would be much the same as that of organogel C18. The length of alkyl chain had not obvious effect on the organogel fluorescence emission.
 |
| | Fig. 6 The optimized geometries of simplified molecule for C6, C12 and C18 in by DFT at the B3LYP/6-31G+(d) level using Gaussian 09. | |
Conclusions
In summary, organic nanofibres and sheet which could gelate some of nonpolar and polar solvents were obtained by self-assembly of a series of simple gelators containing 3,3′,4,4′-biphenyltetracarboxylic. The morphology and surface wettability of the gel could be tuned according to the length of alkyl chain changing. The morphology of the gel in petroleum ether changed from nanofibres to sheet with the length of alkyl chain increasing. The structure surface hydrophobic properties of the gel in petroleum ether were decreased via increasing the length of alkyl chain. Especially, the superhydrophobic surface was obtained from organogel C12 which had the effect of the lotus leaf. There was not any hydrogen bonding in this self-assembly system. So the π–π stacking and van der Waals force were possible the mainly driving force for the gel formation. This kind of superhydrophobic surface is expected to be used in the field of numerous technical applications in resisting water coalescence, fog condensation, and preventing contamination.
Acknowledgements
Cao et al. gratefully acknowledges Prof. Tao Yi of Fudan University for partial experimental support and research fellowships. This work was supported by National Natural Science Foundation of China (21401159), Program for University Innovative Research Team of Henan (2012IRTSHN017, 15IRTSTHN001), and the Science and Technology Key Project of Henan Education Department (13A150760), Project Supported by University Students Sustentation Fund of Xinyang Normal University (2014-DXS-128).
References
- B. N. Sahoo and B. Kandasubramanian, RSC Adv., 2014, 4, 22053–22093 RSC.
- H. Zhu, Z. Guo and W. Liu, Chem. Commun., 2014, 50, 3900–3913 RSC.
- T. Sun, G. Qing and L. Jiang, Chem. Soc. Rev., 2010, 40, 2909–2921 RSC.
- K. Liu and L. Jiang, Nano Today, 2011, 6, 155–175 CrossRef CAS.
- J. Drelich, E. Chibowski, D. D. Meng and K. Terpilowski, Soft Matter, 2011, 7, 9804–9828 RSC.
- Q. Liu, X. Wang, B. Yu, F. Zhou and Q. Xue, Langmuir, 2012, 28, 5845–5849 CrossRef CAS PubMed.
- F. Xiao, S. Yuan, B. Liang, G. Li, S. Pehkonenc and T. Zhang, J. Mater. Chem. A, 2015, 3, 4374–4388 CAS.
- J. Zheng, S. Bao, Y. Guo and P. Jin, ACS Appl. Mater. Interfaces, 2014, 6, 1351–1355 CAS.
- Y. Zhang, Y. Chen, L. Shi, J. Li and Z. Guo, J. Mater. Chem., 2012, 22, 799–815 RSC.
- F. Xia, Y. Zhu, L. Feng and L. Jiang, Soft Matter, 2009, 5, 275–281 RSC.
- P. Wei, X. Yan and F. Huang, Chem. Soc. Rev., 2015, 44, 815–832 RSC.
- Z. Wei, J. Yang, J. Zhou, F. Xu, M. Zrínyi, P. Dussault, Y. Osada and Y. Chen, Chem. Soc. Rev., 2014, 43, 8114–8131 RSC.
- R. G. Weiss, J. Am. Chem. Soc., 2014, 136, 7519–7530 CrossRef CAS PubMed.
- S. S. Babu, V. K. Praveen and A. Ajayaghosh, Chem. Rev., 2014, 114, 1973–2129 CrossRef CAS PubMed.
- V. K. Singh, A. Anis, I. Banerjee, K. Pramanik, M. K. Bhattacharya and K. Pal, Mater. Sci. Eng., C, 2014, 44, 151–158 CrossRef CAS PubMed.
- Q. He, Y. Cui and J. Li, Chem. Soc. Rev., 2009, 38, 2292–2303 RSC.
- J. Wu, T. Yi, T. Shu, M. Yu, Z. G. Zhou, M. Xu, Y. Zhou, H. Zhang, J. Han, F. Li and C. Huang, Angew. Chem., Int. Ed., 2008, 120, 1079–1083 CrossRef.
- X. Cao, J. Zhou, Y. Zou, M. Zhang, X. Yu, S. Zhang, T. Yi and C. Huang, Langmuir, 2011, 27, 5090–5097 CrossRef CAS PubMed.
- Y. Zhou, M. Xu, T. Yi, S. Xiao, Z. Zhou, F. Li and C. Huang, Langmuir, 2007, 23, 202–208 CrossRef CAS PubMed.
- M. Yamanaka, K. Sada, M. Miyata, K. Hanabusad and K. Nakano, Chem. Commun., 2006, 2248–2250 RSC.
- X. Yang, G. Zhang, D. Zhang, J. Xiang, G. Yang and D. Zhu, Soft Matter, 2011, 7, 3592–3598 RSC.
- Y. Yan, X. Wang, J. I. L. Chen and D. S. Ginger, J. Am. Chem. Soc., 2013, 135, 8382–8387 CrossRef CAS PubMed.
- T. Ogoshi, K. Kida and T. Yamagishi, J. Am. Chem. Soc., 2012, 134, 20146–20150 CrossRef CAS PubMed.
- C. Wang, Q. Chen, F. Sun, D. Zhang, G. Zhang, Y. Huang, R. Zhao and D. Zhu, J. Am. Chem. Soc., 2010, 132, 3092–3096 CrossRef CAS PubMed.
- H. Koshima, N. Ojima and H. Uchimoto, J. Am. Chem. Soc., 2009, 131, 6890–6891 CrossRef CAS PubMed.
- P. Bairi, B. Roy, P. Routh, K. Sen and A. K. Nandi, Soft Matter, 2012, 8, 7436–7445 RSC.
- S. Yagai, T. Seki, T. Karatsu, A. Kitamura and F. Wurthner, Angew. Chem., Int. Ed., 2008, 47, 3367–3371 CrossRef CAS PubMed.
- N. C. Maiti, S. Mazumdar and N. Periasamy, J. Phys. Chem. B, 1998, 102, 1528–1538 CrossRef CAS.
- P. Bairi, B. Roy and A. K. Nandi, J. Phys. Chem. B, 2010, 114, 11454–11461 CrossRef CAS PubMed.
- X. H. Cao, L. Y. Meng, Z. H. Li, Y. Y. Mao, H. C. Lan, L. M. Chen, Y. Fan and T. Yi, Langmuir, 2014, 30, 11753–11760 CrossRef CAS PubMed.
- T. Sun, L. Feng, X. Gao and L. Jiang, Acc. Chem. Res., 2005, 38, 644–652 CrossRef CAS PubMed.
- L. Feng, S. Li, Y. Li, H. Li, L. Zhang, J. Zhai, Y. Song, B. Liu, L. Jiang and D. Zhu, Adv. Mater., 2002, 14, 1857–1860 CrossRef CAS.
- M. J. Liu, Y. M. Zheng, J. Zhai and L. Jiang, Acc. Chem. Res., 2010, 43, 368–377 CrossRef CAS PubMed.
- H. Shao, J. Seifert, N. C. Romano, M. Gao, J. J. Helmus, C. P. Jaroniec, D. A. Modarelli and J. R. Parquette, Angew. Chem., Int. Ed., 2010, 49, 7688–7691 CrossRef CAS PubMed.
- H. Shao, M. Gao, S. H. Kim, C. P. Jaroniec and J. R. Parquette, Chem.–Eur. J., 2011, 17, 12882–12885 CrossRef CAS.
- M. Zhang, B. Wang, T. Jiang, M. Jiang and T. Yi, CrystEngComm, 2012, 14, 8057–8062 RSC.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785–789 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra05841d |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 






 in organogel C6, indicated a more complicated structure (Fig. S7b†). There were few peaks for organogel C12 and C18. Insight into the molecule structure in gel state, the optimized conformation of the truncated models (alkyl chains are replaced by Me groups) of those compounds in monomer was gained using the hybrid B3LYP density functional theory method with the 6-31+G(d) basis set.37,38 Optimized structures (B3LYP/6-31G+(d)) was showed in Fig. 6. The dihedral angle of the two benzene rings of the molecule C6, C12 and C18 was 39.3°. This result well explained the difference of fluorescence emission of organogels C6, C12 and C18 in petroleum ether. If the two benzene rings were in a plane, then the fluorescence emission of organogels C6 and C12 would be much the same as that of organogel C18. The length of alkyl chain had not obvious effect on the organogel fluorescence emission.
in organogel C6, indicated a more complicated structure (Fig. S7b†). There were few peaks for organogel C12 and C18. Insight into the molecule structure in gel state, the optimized conformation of the truncated models (alkyl chains are replaced by Me groups) of those compounds in monomer was gained using the hybrid B3LYP density functional theory method with the 6-31+G(d) basis set.37,38 Optimized structures (B3LYP/6-31G+(d)) was showed in Fig. 6. The dihedral angle of the two benzene rings of the molecule C6, C12 and C18 was 39.3°. This result well explained the difference of fluorescence emission of organogels C6, C12 and C18 in petroleum ether. If the two benzene rings were in a plane, then the fluorescence emission of organogels C6 and C12 would be much the same as that of organogel C18. The length of alkyl chain had not obvious effect on the organogel fluorescence emission.
