
A general and practical route to 4,5-disubstituted oxazoles using acid chlorides and isocyanides†
Dipak Kumar Tiwari,
Jaya Pogula and
Dharmendra Kumar Tiwari*
Inorganic and Physical Chemistry Division, CSIR-Indian Institute of Chemical Technology, Tarnaka, Hyderabad-500007, India. E-mail: dktiwari@iict.res.in; dkt80.org@gmail.com
First published on 4th June 2015
Abstract
An efficient and mild method for the synthesis of 4,5-disubstituted oxazoles has been developed. A series of 4,5-disubstituted oxazoles were prepared via [3 + 2] cycloaddition reaction of various isocyanides and acid chlorides in the presence of base. This method serves as an efficient preparation of 5-formyloxazole which might prove to be a synthetically useful intermediate.
Introduction
Oxazoles represent an important class of five membered heterocycles, occuring as substructural units in a wide variety of biologically active compounds1 (diazonamide A (1)1i and leucamide A (2)1j), pharmaceuticals2 (ostreogrycin-A (3) Fig. 1) and molecular sensors.3 Oxazoles are also popular as valuable precursors in many useful synthetic transformations.4 In recent years, 4,5-disubstituted oxazoles have received considerable attention from both synthetic and medicinal chemists, mainly due to their significant therapeutic potential in treating various diseases such as inflammation, cancer and asthma.5,6 4,5-Disubstituted oxazoles as a class are a potent and selective inhibitor of the stress-activated kinase p38α,7 cyclooxygenate-II (COX-II), cyclooxygenase-1 (COX-1) and anti-tumor necrosis factor (anti-TNF).8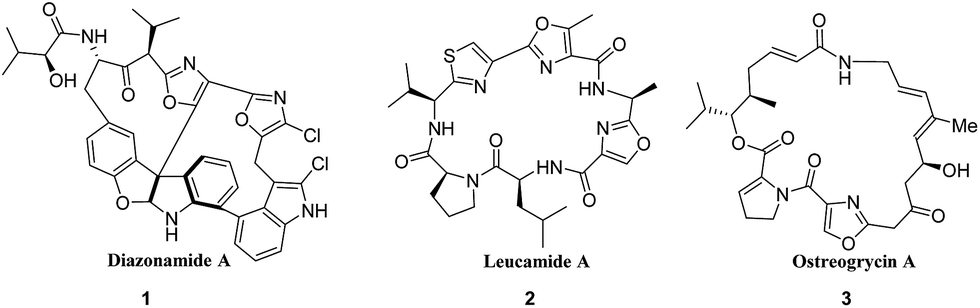 | ||
| Fig. 1 Oxazole containing biologically active compounds. | ||
Owing to their medicinal value, a number of synthetic methods have been developed to construct 4,5-disubstituted oxazoles.9
Among these methods, the most classical procedure is the cyclocondensation of acetyl chloride and monosubstituted tosyl methyl isocyanides developed by Van Leusen et.al. (Fig. 2 eqn (1)).10a Later, Yu and Stevens groups reported improved syntheses in ionic liquids and an automated flow reactor system respectively.10b,c In addition, Miller group devised a new route which involves the regioselective C2 silylation followed by metalation of oxazoles at C5 position (Fig. 2 eqn (2)).10d
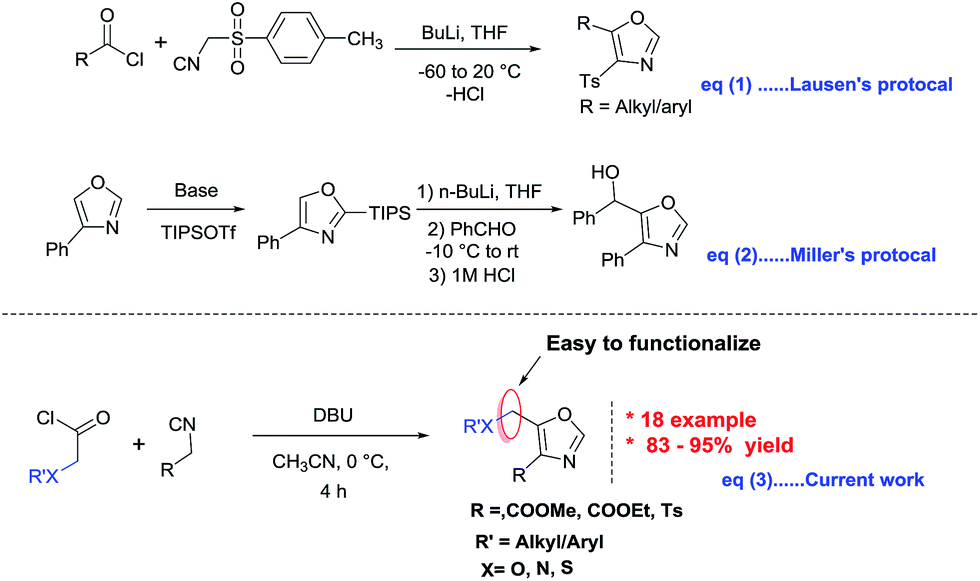 | ||
| Fig. 2 Scheme depicting previous reports on the synthesis of 4,5-disubstituted oxazoles along with present study. | ||
While all these useful methods provide convenient access to 4,5-disusbtituted oxazoles, there are certain limitations associated with them, such as harsh reaction conditions, multistep synthesis, and difficulties in further functionalization, especially at C5 position of oxazoles. Therefore, development of a mild and efficient approach to 4,5-disubstituted oxazoles is still highly desirable.
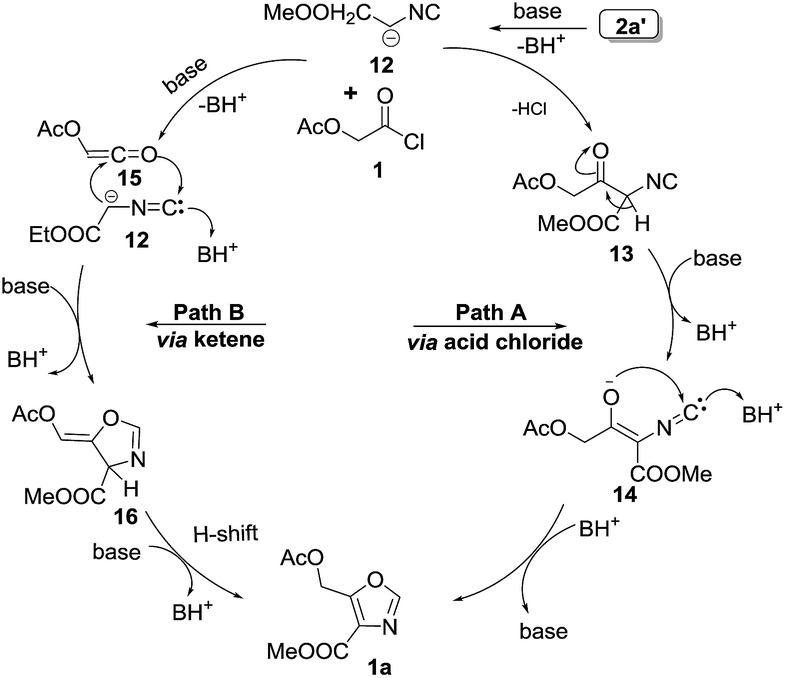 | ||
| Fig. 3 Mechanism of [3 + 2] cycloaddition of acid chloride and isocyanide towards 4,5-disubstituted oxazoles. | ||
Acid chlorides have long been renowned for their reactivity in numerous synthetic transformations, particularly their reactivity in [2 + 2] and [4 + 2] cycloadditions, reductive couplings, SN2 substitutions as well as both nucleophilic and electrophilic additions. Our current research is focused on the synthesis of various azetidin-2-ones using [2 + 2] cycloaddition reactions of imines with in situ generated ketenes from acid chlorides.11 As a continuation of the same research program which uses acid chlorides as precursors, herein we wish to report an efficient and practical synthesis of 4,5-disubstituted oxazole via [3 + 2] cycloaddition reactions of isocyanides with alkyloxy/aryloxy acid chlorides in the presence of a base (Fig. 2 eqn (3)). This methodology works with broad spectrum of acid chlorides including the ones containing heteroatoms such as oxygen, nitrogen and sulphur. These 4,5-disubstituted oxazoles could in turn be functionalized at C5 position.
Results and discussion
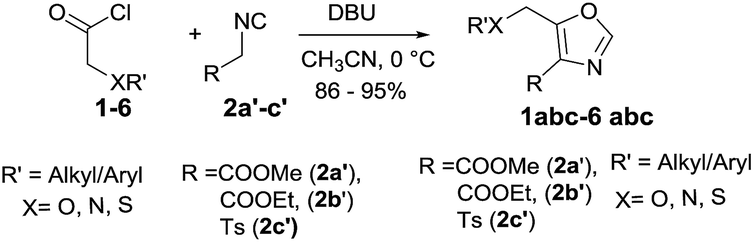
Initially, the synthesis of 4,5-disubstituted oxazole was examined using acetoxyacetyl chloride 1 and methyl 2-isocyanoacetate 2a′ as the model substrate. Our first attempt by treating acetoxyacetyl chloride 1 (0.5 mmol) and methyl 2-isocyanoacetate 2a′ (0.5 mmol) in presence of anhydrous K2CO3 (2.0 equiv.) in dry DCM at room temperature was unsuccessful (Table 1 entry-1). Use of different bases such as Cs2CO3, Et3N and DBU in dichloromethane too failed to yield the desired product 1a (Table 1 entries 1–4). However, 20% yield of the product 4,5-disubstituted oxazole 1a was isolated when reaction was performed in anhydrous CH3CN in the presence of K2CO3 (2.0 equiv.) at room temperature under argon atmosphere (Table 1, entry 5). Furthermore, 1a was isolated in 35% yield by using 2.0 equiv. of Et3N in CH3CN at room temperature (Table 1, entry-6). The poor performance of the reactions at room temperature, possibly may be due to the polymerization of ketene generated in situ from acid chloride.12 Therefore, we attempted reaction at lower temperature and a sharp increase in the yield (53%) of 1a was observed when reaction was performed at 0 °C (Table 1, entry 7). To our delight, a much better yield (76%) of 1a was obtained when DBU (2.0 equiv.) was employed as a base (Table 1, entry-8). Notably, the conversion and the yields were affected dramatically by amount of DBU employed, and the best result (95%) was obtained by employing 3.5 equiv. of DBU (Table 1, entry 9). Further increase or decrease in the amount of DBU did not give a better yield (Table 1, entry 10 & 11). Stronger bases such as KOH, NaOMe and KOtBu were found less effective to promote the reaction (Table 1, entries 12–14) and furnished lower yields of desired 1a. It was plausibly due the enolization of acetate group of the substrate (1) and product (1a) in the presence of strong bases which led to the formation of undesired side products. However, strong bases were equally effective as DBU with substrates containing no acetate group (For details, see ESI†). Furthermore, the cycloaddition reaction of 1 and 2a′ were tried in different solvents such as N,N dimethyl formamide (DMF), dimethylsulfoxide (DMSO), tetrahydrofuran (THF) and 1,4-dioxane but most of them were found less efficient, resulting 1a in inferior yields (Table 1, entries 15–18). Unsurprisingly, no product was obtained in protic solvents such as MeOH and EtOH (entries 19 & 20).
|
|||||
|---|---|---|---|---|---|
| Entry | Base/equiv. | Solvent | Temp. °C | Time (h) | Yieldb (%) |
| a All reactions were carried out on 0.5 mmol of acetoxyacetyl chloride (1), 0.5 mmol of methylisocyanoacetate (2a′).b Isolated yield. | |||||
| 1 | K2CO3 (2) | DCM | rt | 6 | No reaction |
| 2 | Cs2CO3 (2) | DCM | rt | 6 | No reaction |
| 3 | Et3N (2) | DCM | rt | 6 | No reaction |
| 4 | DBU (2) | DCM | rt | 6 | No reaction |
| 5 | K2CO3 (2) | CH3CN | rt | 6 | 20% |
| 6 | Et3N (2) | CH3CN | rt | 6 | 35% |
| 7 | Et3N (2) | CH3CN | 0 °C | 6 | 53% |
| 8 | DBU (2) | CH3CN | 0 °C | 6 | 76% |
| 9 | DBU (3.5) | CH3CN | 0 °C | 4 | 95% |
| 10 | DBU (3.0) | CH3CN | 0 °C | 4 | 84% |
| 11 | DBU (4.0) | CH3CN | 0 °C | 4 | 91% |
| 12 | KOH (3.5) | CH3CN | 0 °C | 4 | 25% |
| 13 | NaOMe (3.5) | CH3CN | 0 °C | 4 | 41% |
| 14 | t-BuOK (3.5) | CH3CN | 0 °C | 4 | 36% |
| 15 | DBU (3.5) | DMSO | 0 °C | 4 | 45% |
| 16 | DBU (3.5) | DMF | 0 °C | 4 | 32% |
| 17 | DBU (3.5) | THF | 0 °C | 4 | 65% |
| 18 | DBU (3.5) | 1,4-Dioxane | 0 °C | 4 | 51% |
| 19 | DBU (3.5) | MeOH | 0 °C | 4 | No reaction |
| 20 | DBU (3.5) | EtOH | 0 °C | 4 | No reaction |
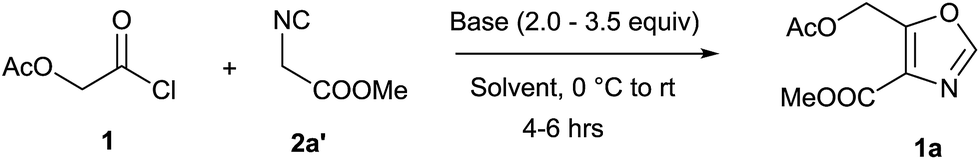
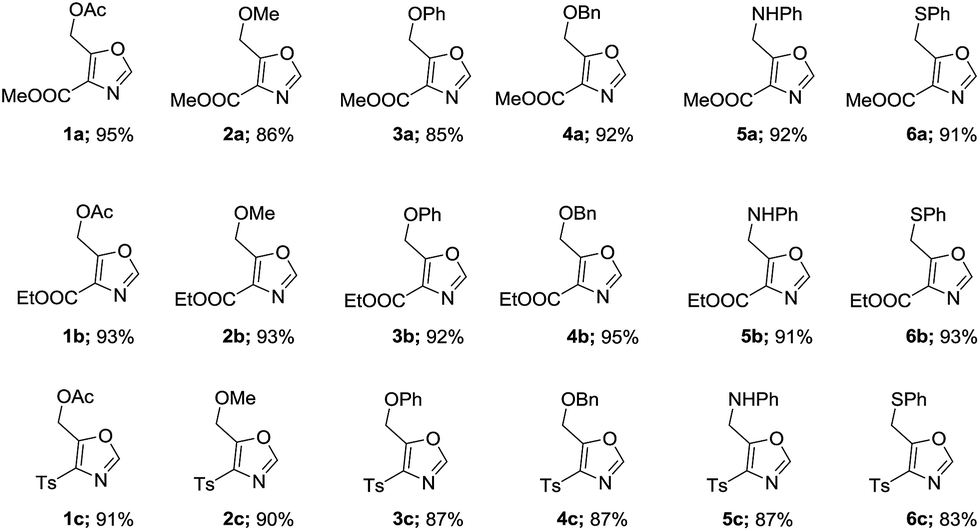
With optimal conditions in hand (Table 1, entry 9), the scope of the reaction with respect to various acid chlorides and isocyanide components was investigated. Methyl 2-isocyanoacetate effectively reacted with acid chlorides bearing both aliphatic and aromatic groups. Methoxyacetyl chloride 2, phenoxy and benzyloxyacetyl chlorides (3 & 4) also gave corresponding 4,5-disubstituted oxazoles 2a, 3a, and 4a in good yields. Nitrogen containing acid chlorides (PhthNCH2COCl, 5) on treatment with methyl 2-isocyanoacetate (2a′) produced corresponding oxazole 5a in 92% yield.
Identical yield (91%) of 6a was obtained when thiophenylacetyl chloride (6) was subjected to similar reaction condition. Also worthy of note is the observation that in place of methyl 2-isocyanoacetate (2a′) both ethyl 2-isocyanoacetate (2b′) and tosylmethyl isocyanide (TosMIC, 2c′) readily participated in cycloaddition with various acid chlorides to give corresponding oxazoles (1b, 1c-5b, 1c) in very good yields (Table 2).
This initial success propelled us to investigate the versatility of present method. 5-formyl oxazole 8 which can be considered to be an intermediate of high utility can undergo various reactions such as Grignard, Aldol, Wittig, and reductive amination to give functionalized oxazoles. Similar 5-formyl oxazole have already been used as an advance intermediate in the preparation of biologically active compound which is known to posses CRTH2 receptor activity.13 This synthetically important intermediate was easily prepared in two step process from 1c (Scheme 1). The hydrolysis of acetate group of 1c using aqueous sodium bicarbonate solution in MeOH gave intermediate 7 which underwent smooth Swern oxidation to furnish 5-formyl-4-tosyl oxazole 8 in 87% yield.
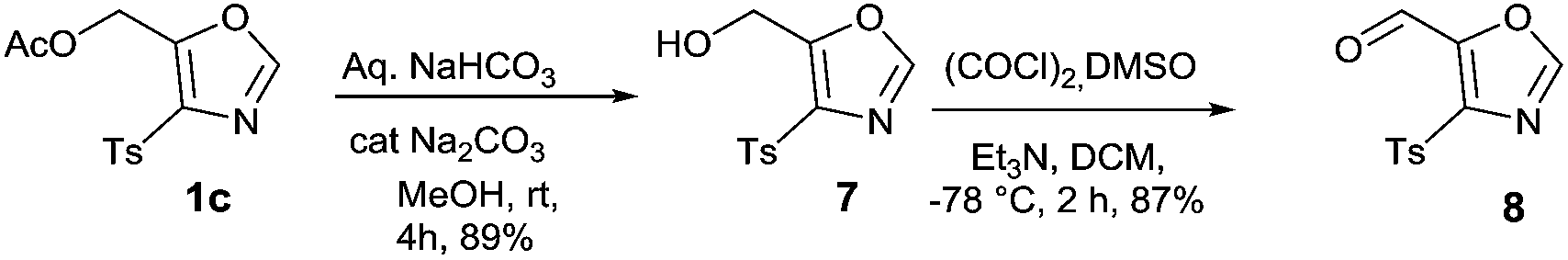 | ||
| Scheme 1 synthesis of 5-formyl oxazole from 1c. | ||
Next, in order to expand the scope of the present methodology, we examined the reaction of aryl cyanides (9a, 9b) with methyl 2-isocyanoacetate 2a′ under standard conditions (Scheme 2) to prepare 4,5-disubstituted imidazole 10.14 But, unfortunately, no desired products (10a,b) were observed. However, when the reaction mixture was refluxed the cyclodimerization of isocyanide takes place to form 11 in 95% yield along with unreacted aryl cyanides (9a,b). The spectral data of 11 is in good agreement with previously reported value.15
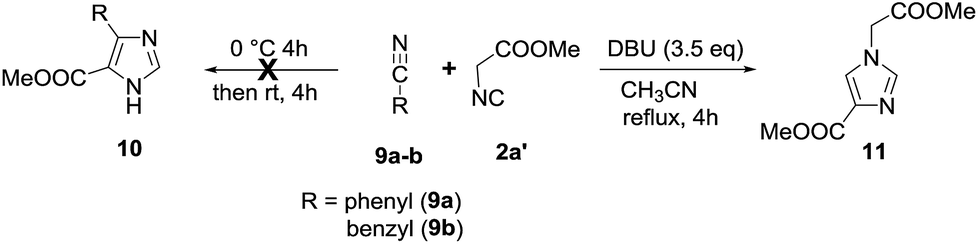 | ||
| Scheme 2 reaction of aryl cyanides (9a,b) with isocyanide (2a). | ||
The formation of 4,5-disubstituted oxazoles may be explained by two pathways (Fig. 3). In pathway A, initially anionic form of 2a′, undergoes addition on acid chloride 1 give intermediate 13. This intermediate 13 then undergoes enolization, followed by nucleophilic attack on carbene form of 14 leading to the formation of 4,5-disubstituted oxazole 1a. Another possible mechanistic pathway could be via ketene mechanism. The ketene 15 generated in situ from the acid chloride undergoes a [3 + 2] cycloaddition with 12 to yield intermediate 16 which undergoes 1,3 H-shift to give 1a.
Conclusions
In summary, 4,5-disubstituted oxazole construction was achieved in very good yields from readily available acid chlorides and isocyanides under very mild conditions. Moreover, this method was applied to the synthesis of 5-formyl oxazole. Work to exploit the potential of 5-formyl oxazoles for further functionalization to yield valuable intermediates is underway and findings will be disclosed in due course of time. The ready availability of starting materials, broad substrate scope, and operational simplicity makes the present method an attractive option for the synthesis of the oxazole skeleton.Experimental
General
The reagents, chemicals and solvents were either purchased from commercial suppliers or prepared and purified by standard techniques. Column chromatography was carried out using silica gel 100–200 mesh. Infrared spectra were recorded using a FT-IR spectrophotometer and values reported in cm−1. 1H and 13C NMR spectra were recorded with 300, 400 and 500 MHz NMR instruments with tertramethylsilane (TMS) as an internal standard. High-resolution mass spectra (ESI-HRMS) were recorded on ESI-QTOP mass spectrometer.Typical experimental procedure for the synthesis of 4,5-disubstituted oxazoles
A solution of acid chlorides (0.5 mmol) in dry CH3CN (2 mL) was slowly added to a mixture of isocyanides (0.5 mmol) and DBU (1.75 mmol) in dry CH3CN (4 mL) at 0 °C. The reaction mixture was allowed to stir for additional 4 h at same temperature. The reaction mixture was quenched with water (6 mL) and the mixture was diluted with ethylacetate (10 mL). The organic layer was separated and washed with aqueous NaHCO3 solution (5 mL) and brine (5 mL). The organic layer was dried over anhydrous sodium sulphate and solvent was removed under reduced pressure to produce crude 4,5-disubstituted oxazole, which was on purification using 100–200 mesh silica gel, gave pure 4,5-disubstituted oxazoles in very good yields.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 541, 1593, 1506, 1323, 1030 cm−1; 1H NMR (300 MHz, CDCl3) 7.92 (d, J = 8.3 Hz, 2H), 7.81 (s, 1H), 7.36 (d, J = 8.0 Hz, 2H), 4.99 (s, 2H), 3.35 (bs, 1H), 2.43 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 154.83, 150.27, 145.51, 136.99, 136.18, 130.07, 128.19, 54.64, 21.72; HRMS (ESI, Orbitrap): calcd for C11H11NO4SNa [M + Na]+ 276.03010 found 276.03018.
541, 1593, 1506, 1323, 1030 cm−1; 1H NMR (300 MHz, CDCl3) 7.92 (d, J = 8.3 Hz, 2H), 7.81 (s, 1H), 7.36 (d, J = 8.0 Hz, 2H), 4.99 (s, 2H), 3.35 (bs, 1H), 2.43 (s, 3H); 13C NMR (75 MHz, CDCl3) δ 154.83, 150.27, 145.51, 136.99, 136.18, 130.07, 128.19, 54.64, 21.72; HRMS (ESI, Orbitrap): calcd for C11H11NO4SNa [M + Na]+ 276.03010 found 276.03018.Acknowledgements
The authors thank the Department of Science and Technology (DST) New Delhi, India, for financial support. The authors are grateful to the Director, CSIR-IICT for providing the necessary infrastructure. D. K. T. is grateful to Dr P. M. Chincholkar for his valuable suggestions.References
- (a) V. S. C. Yeh, Tetrahedron, 2004, 60, 11995 CrossRef CAS PubMed; (b) J. R. Lewis, Nat. Prod. Rep., 2002, 19, 223 RSC; (c) P. Wipf, Chem. Rev., 1995, 95, 2115 CrossRef CAS; (d) Z. Jin, Nat. Prod. Rep., 2003, 20, 584 RSC; (e) Z. Jin, Z. Li and R. Huang, Nat. Prod. Rep., 2002, 19, 454 RSC; (f) Z. Jin, Nat. Prod. Rep., 2009, 26, 382 RSC; (g) E. Riego, D. Hernandez, F. Albercia and M. Alvarez, Synthesis, 2005, 1907 CrossRef CAS PubMed; (h) Z. Jin, Nat. Prod. Rep., 2011, 28, 1143 RSC; (i) J. Li, A. W. G. Brugett, L. Esser, C. Amezcua and P. G. Harran, Angew. Chem., Int. Ed., 2001, 40, 4770 CrossRef CAS; (j) S. Kehraus, G. M. Konig and A. D. Wright, J. Org. Chem., 2002, 67, 4989 CrossRef CAS PubMed.
- (a) P. Wipf and F. Yokokawa, Tetrahedron Lett., 1998, 39, 2223 CrossRef; (b) A. Nishida, M. Fuwa, S. Naruto, Y. Sugano, H. Saito and M. Nakagawa, Tetrahedron Lett., 2000, 41, 4791 CrossRef CAS; (c) G. R. Delpierre, F. W. Eastwood, G. E. Gream, D. G. I. Kingston, P. S. Sarin, L. Todd and D. H. Williams, J. Chem. Soc. C, 1966, 165, 3–1669 Search PubMed; (d) G. R. Delpierre, F. W. Eastwood, G. E. Cream, D. G. I. Kingston, P. S. Sarin, L. Todd and D. H. Williams, Tetrahedron Lett., 1966, 36, 9–372 Search PubMed.
- (a) A. Tarraga, P. Molina, D. Curiel and M. D. Velasco, Organomettalics, 2001, 20, 2145 CrossRef CAS; (b) A. Tarraga, P. Molina, D. Curiel and M. D. Velasco, Tetrahedron, 2001, 57, 6765 CrossRef CAS.
- (a) F. W. Hartner, Comprehensive Heterocyclic Chemistry II, ed. Katritzky A. R., Rees C. W., Elsevier, Oxford, 1996, vol. 3, p. 261 Search PubMed; (b) J. R. Lewis, Nat. Prod. Rep., 2001, 18, 95 RSC; (c) G. V. Boyd, Prog. Heterocycl. Chem., 1999, 11, 213 CAS; (d) R. Jansen, H. Irschik, H. Reichenbach, V. Wray and G. Hoefle, Liebigs Ann. Chem., 1994, 759 CrossRef CAS PubMed; (e) I. V. Hartung, B. Niess, L. O. Haustedt and H. M. R. Hoffmann, Org. Lett., 2002, 4, 3239 CrossRef CAS PubMed; (f) C.-M. Cheung, F. W. Goldberg, P. Magnus, C. J. Russell, R. Turnbull and V. Lynch, J. Am. Chem. Soc., 2007, 129, 12320 CrossRef CAS PubMed; (g) T. E. Smith, W.-H. Kuo, E. P. Balskus, V. D. Bock, J. L. Roizen, A. B. Theberge, K. A. Carroll, T. Kurihara and J. D. Wessler, J. Org. Chem., 2008, 73, 142 CrossRef CAS PubMed.
- (a) S. A. Lipton, PCT Int. Appl. WO 0101986, 2001; (b) M. Andersson, L. Hansen Peter, A. Nikitidis and P. Sjoelin, PCT Int. Appl. WO 2005026123, 2005, Indian Chem. Abstr., 142, 316705 Search PubMed; (c) T. Kline, J. Bowman, B. H. Iglewski, T. De Kievit, Y. Kakai and L. Passador, Bioorg. Med. Chem. Lett., 1999, 9, 3447 CAS; (d) R. Laszo and A. Schlapbach, PCT Int. Appl. WO 0063204, 2000, Indian Chem. Abstr., 133, 321897 Search PubMed; (e) L. Revesz, E. Blum, F. E. Di Padova, T. Buhl, R. Feifel, H. Gram, P. Hiestand, U. Manning and G. Rucklin, Bioorg. Med. Chem. Lett., 2004, 14, 3595 CrossRef CAS PubMed; (f) L. Revesz and A. Schlapbach. PCT Int. Appl. WO 2000063204, 2000, Indian Chem. Abstr., 133, 321897 Search PubMed.
- (a) L. C. Blumberg and M. J. Munchhof. PCT Int. Appl. WO 2004026863, 2004, Indian Chem. Abstr., 140, 287371 Search PubMed; (b) N. Svenstrup, A. Kuhl, D. Flubacher, M. Brands, K. Ehlert, C. Ladel, M. Otteneder and J. Keldenich. PCT Int. Appl. WO 2003072574, 2003, Indian Chem. Abstr., 139, 230786 Search PubMed; (c) L. Alcaraz, M. Furber, M. Purdie and B. Springthorpe. PCT Int. Appl. WO 2003068743, 2003, Indian Chem. Abstr., 139, 197375 Search PubMed; (d) J. L. Adams, T. F. Gallapher, J. C. Boehm and S. M. Thompson. PCT Int. Appl. WO 9513067, 1995, Indian Chem. Abstr., 123, 152881 Search PubMed.
- (a) K. F. McClure, M. A. Letavic, A. S. Kalgutkar, C. A. Gabel, L. Audoly, J. T. Barberia, J. F. Braganza, D. Carter, J. Thomas, T. J. Carty, S. R. Cortina, M. A. Dombroski, K. M. Donahue, N. C. Elliott, C. P. Gibbons, C. K. Jordan, A. V. Kuperman, J. M. Labasi, R. E. LaLiberte, J. M. McCoy, B. M. Naiman, K. L. Nelson, H. T. Nguyen, K. M. Peese, F. J. Sweeney, T. J. Taylor, C. E. Trebino, Y. A. Abramov, E. R. Laird, W. A. Volberg, J. Zhou, J. Bach and F. Lombardo, Bioorg. Med. Chem. Lett., 2006, 16, 4339 CrossRef CAS PubMed; (b) R. A. Buzon, M. J. Castaldi, Z. B. Li, D. H. B. Ripin and Y. Tao. PCT Int. Appl. WO 2004020438, 2004, Indian Chem. Abstr., 140, 235721 Search PubMed; (c) K. F. McClure, Y. A. Abramov, E. R. Laird, J. T. Barberia, W. Cai, T. J. Carty, S. R. Cortina, D. E. Danley, A. J. Dipesa, K. M. Donahue, M. A. Dombroski, N. C. Elliott, C. A. Gabel, S. Han, T. R. Hynes, P. K. LeMotte, M. N. Mansour, E. S. Marr, M. A. Letavic, J. Pandit, D. B. Ripin, F. J. Sweeney, D. Tan and Y. J. Tao, Med. Chem., 2005, 48, 5728 CrossRef CAS PubMed.
- (a) J. B. Doherty, J. E. Stelmach and M.-H. Chen, Expert Opin. Ther. Pat., 2003, 13, 381 CrossRef CAS; (b) J. Westra, M. Doornbos-van der, P. de Boer, M. A. van Leeuwen, M. H. van Rijswijk and P. C. Limburg, Arthritis Res. Ther., 2004, 6, R384 CrossRef CAS PubMed.
- (a) B. Li, R. A. Buzon and Z. J. Zhang, Org. Process Res. Dev., 2007, 11, 951 CrossRef CAS; (b) S. Brahma and J. K. Ray, J. Heterocycl. Chem., 2008, 45, 311 CrossRef CAS PubMed; (c) J. R. Davis, P. D. Kane and C. J. Moody, Tetrahedron, 2004, 60, 3967 CrossRef PubMed; (d) M. Ohba, H. Kubo, S. Seto, T. Fujii and H. Ishibashi, Chem. Pharm. Bull., 1998, 46, 860 CrossRef CAS; (e) E. Vedejs and L. M. Luchetta, J. Org. Chem., 1999, 64, 101 CrossRef PubMed; (f) D. R. Williams and E. L. MeClymont, Tetrahedron Lett., 1993, 34, 7705 CrossRef CAS.
- (a) A. M. Van Leusen, B. E. Hoogenboom and H. Siderius, Tetrahedron Lett., 1972, 23, 2369 CrossRef; (b) B. Wu, J. Wen, J. Zhang, J. Li, Y.-Z. Xiang and X. Q. Yu, Synlett, 2009, 500 CAS; (c) M. Baumann, I. R. Baxendale, S. V. Ley, C. D. Smith and G. K. Tranmer, Org. Lett., 2006, 8, 5231 CrossRef CAS PubMed; (d) R. A. Miller, R. M. Smith and B. Marcune, J. Org. Chem., 2005, 70, 9074 CrossRef CAS PubMed.
- (a) D. K. Tiwari, A. Y. Shaikh, S. L. Pavase, V. K. Gumaste and A. R. A. S. Deshmukh, Tetrahedron, 2007, 63, 2524 CrossRef CAS PubMed; (b) D. K. Tiwari, V. K. Gumaste and A. R. A. S. Deshmukh, Synthesis, 2005, 115 Search PubMed; (c) G. Pandey and D. K. Tiwari, Tetrahedron Lett., 2009, 50, 3296 CrossRef CAS PubMed; (d) D. K. Tiwari, K. C. Bharadwaj, V. G. Puranik and D. K. Tiwari, Org. Biomol. Chem., 2014, 12, 7389 RSC.
- (a) F. A. Leibfarth, M. Kang, M. Ham, J. Kim, L. M. Campos, N. Gupta, B. Moon and C. J. Hawker, Nature Chemistry, 2010, 2, 207–212 CrossRef CAS PubMed; (b) U. Salzner and S. M. Bachrach, J. Am. Chem. Soc., 1994, 116, 6850–6855 CrossRef CAS; (c) L. G. Drayton and H. W. J. Thompson, J. Chem. Soc., 1948, 1416–1419 RSC.
- H. George, M. J. Gary, H. Finch, A. Rosa and A. Shahadat, PCT Int. Appl. WO 2010142934 A1 20101216. 2010.
- T. Murakami, M. Otsuka and M. Ohno, Tetrahedron Lett., 1982, 23, 4729–4732 CrossRef CAS.
- (a) C. Kanazawa, S. Kamijo and Y. Yamamoto, J. Am. Chem. Soc., 2006, 128, 10662–10663 CrossRef CAS PubMed; (b) R. Grigg, M. I. Lansdell and M. Tornton-Pett, Tetrahedron, 1999, 55, 2025–2044 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07668d |
| This journal is © The Royal Society of Chemistry 2015 |