
Continuous-flow hydrogenation of olefins and nitrobenzenes catalyzed by platinum nanoparticles dispersed in an amphiphilic polymer†
Abstract
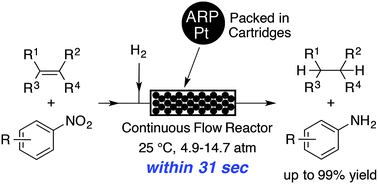
A method for the flow hydrogenation of olefins and nitrobenzenes in a continuous-flow reactor containing platinum nanoparticles dispersed on an amphiphilic polystyrene–poly(ethylene glycol) resin (ARP-Pt) was developed. The hydrogenation of olefins and nitrobenzenes was completed within 31 seconds in the continuous-flow system containing ARP-Pt, giving the corresponding hydrogenated products in up to 99% yield with good chemoselectivity. Moreover, long-term (63–70 h) continuous-flow hydrogenation of styrene and nitrobenzene produced more than ten grams of ethylbenzene and aniline, respectively, without significant loss of catalytic activity. The flow hydrogenation system provides an efficient and practical method for the chemoselective reduction of olefins and nitrobenzenes.
 Please wait while we load your content...
Please wait while we load your content...

