DOI:
10.1039/C5RA07497E
(Paper)
RSC Adv., 2015,
5, 55550-55560
Unzipped carbon nanotubes: analytical and binding applications of semisynthetic phlebotropic flavonoid, diosmin†
Received
24th April 2015
, Accepted 16th June 2015
First published on 17th June 2015
Abstract
The present study describes the utility of unzipped carbon nanotubes as electrochemical sensing platforms for the determination of diosmin (DIO) in pharmaceutical formulations and for the investigation of its binding to the drug transport protein, human serum albumin (HSA). Graphene oxide nanoribbons (GONRs) were prepared by unzipping of multiwalled carbon nanotubes and characterized by employing powder XRD, FTIR, absorption, Raman, AFM, SEM and electrochemical impedance spectroscopic methods. The suspension of GONRs was drop cast onto a glassy carbon electrode (GCE) and then was subjected to electrochemical reduction (Er) in the potential range of 0.8 to −1.6 V to obtain ErGONRs/GCE. DIO showed a redox peak (Epa = 0.587 V and Epc = 0.548 V) and an irreversible oxidation peak at 0.845 V on ErGONRs/GCE in a phosphate buffer of pH 3. DIO exhibited an enhanced electrochemical response (∼39-fold increment in the peak current) at ErGONRs/GCE when compared to that at bare GCE. Linearity between the peak current and concentration of DIO was noticed in the range of 51.01–39.21 μM and 25–3.48 μM for differential pulse and square wave voltammetric methods, respectively. The practical utility of the proposed sensor was established by determining DIO in pharmaceutical formulations. Furthermore, the sensor was used for understanding the binding mechanism of the DIO–HSA system. The binding constant and binding ratio between HSA and DIO were calculated to be 2.58 × 104 M−1 and 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, respectively.
:1, respectively.
1. Introduction
Graphene nanoribbons (GNRs) have been recognized as promising building blocks for nanoelectronic and spintronic devices in the carbon family.1–6 The interest in this material has grown exponentially after free-standing graphene layers were unexpectedly found and then follow-up experiments confirmed their massless Dirac fermion charge carrier abilities.7–9 It is desirable to produce high-quality GNRs with well-defined widths and edge structures to explore their applications.10 Several methods have been reported for the preparation of GNRs. In particular, the synthesis of GNRs by unzipping of carbon nanotubes (CNTs) has received much attention because it is possible to produce GNRs with controllable widths, edge structures and layer numbers. The methods of unzipping of CNTs include Ar plasma etching,11 sonochemical unzipping12,13 oxidation cutting,14,15 metal catalytic cutting,16 intercalation and exfoliation.17 Apart from these methods, a simple and most successful solution-based high yield protocol was adopted in recent years for the synthesis of GNRs.18 This protocol involves two steps viz., (i) the oxidative unzipping of CNTs by the application of a strong oxidizing agent (KMnO4) to obtain graphene oxide nanoribbons (GONRs) and (ii) the reduction of GONRs by chemical19 thermal20 or electrochemical21 treatment. Of these reduction approaches, reagent-free electrochemical method is increasingly employed due to its simplicity and eco-friendly nature.22 In view of this, it was planned to prepare a new sensing interface based on electrochemically reduced GONRs for analytical and binding applications.
Diosmin (DIO), chemically known as [3′,5,7-trihydroxy-4′-methoxyflavone-7-rutinoside], is a typical semi-synthetic phlebotropic flavonoid. It is found in a variety of citrus fruits. It inhibits chemically induced cancers in rodents including N-methyl-N-amylnitrosamine-induced esophageal cancer, 4-nitroquinoline 1-oxide-induced oral cancer and azoxymethane-induced colon cancer.23–27 DIO is considered to be a vascular-protecting agent used to treat chronic venous insufficiency, hemorrhoids, lymphedema, varicose veins, anti-inflammatory, free-radical scavenging and antimutagenic properties.28–32
Spectrophotometric,33,34 chromatographic35–40 and electrochemical41–43 methods have been reported for the assay of DIO in pharmaceutical formulations and biological samples. However, the reported chromatographic and spectrophotometric methods require long analysis time, elaborate extraction and purification steps or on-line sample extraction and are relatively costly while the reported electrochemical methods have inherent limitations such as the use of mercury based electrode/gold electrode or less sensitive for the determination of DIO. So, it was thought worthwhile to develop a new electrochemical sensing interface for sensitive determination of DIO.
Interactions between small molecules including bioactive compounds and transport proteins have been widely investigated during last few years.44–49 Serum albumin is one of the most abundant carrier proteins and plays an important role in the transport and disposition of endogenous and exogenous ligands present in the blood.50 Thus, it is significant to study the interaction of bioactive compounds including drugs with proteins. The molecular interactions between human serum albumin (HSA) and some flavonoids have been investigated successfully.51–58 Thus, it is reported that the serum albumin plays a decisive role in the transport and disposition of flavonoids.59 A number of biochemical investigations have revealed that proteins (including enzymes) are frequently the “targets” for therapeutically active flavonoids of both natural and synthetic origin.60 Till now, the use of GNRs based electrochemical sensor or no electrochemical method, was developed for the characterization of binding of DIO–HSA system. Hence, it was thought worthwhile to explore the application of the proposed electrochemical sensor to understand the binding of DIO with HSA.
The present study mainly describes the eco-friendly synthesis and characterization of clean and high-quality 2D GNRs with a length up to 4 μm for electrosensing applications. GNRs with predefined properties were easily and rapidly obtained by oxidative unzipping of MWCNTs. Further, it was followed by electrochemical reduction. These electrochemically reduced (Er) GONRs (ErGONRs) were utilized for the fabrication of electrochemical sensor for analytical applications of DIO. Further, application of the proposed electrochemical sensor was extended for exploring the binding characteristics of DIO with HSA.
2. Experimental
2.1. Reagents and solutions
DIO and HSA were purchased from Sigma Aldrich, USA and used without further purification. A stock solution of 1 mM DIO was prepared by dissolving it in 2 mL NaOH (0.3 M) and then diluting to 10 mL with Millipore water. It was stored in a refrigerator at 4 °C. A stock solution of HSA (250 μM) was prepared in 0.1 M phosphate buffer of pH 7.4 containing 0.15 M NaCl. Working solutions of DIO were prepared by appropriately diluting the stock solution with the selected supporting electrolyte. All other chemicals used were of analytical grade and used as received. Millipore water was used for preparing all solutions.
2.2. Material characterization
X-ray diffraction patterns were recorded on a X-ray diffraction system (Bruker AXS D8) employing the Cu Kα line (1.5406 Å). FTIR and Raman studies were carried out on a NXR-FT-RAMAN, NICOLET, USA and NICOLET 6700, NICOLET, USA respectively. Atomic force microscopy (AFM) images were taken on a Multi-Mode Nanosurf EasyScan atomic force microscope (Nanosurf, Switzerland). Commercially available AFM cantilever tips (Tap190Al-G) with a force constant of 48 N m−1 and a resonance vibration frequency of ∼160 kHz were used. Surface morphologies of GONRs and ErGONRs were performed on a JEOL Model JSM-6390LV scanning electron microscope (SEM).
2.3. Apparatus
Electrochemical measurements were made on a CHI-1103A Electrochemical Analyzer (CH Instruments, Inc. USA) consisting of a GCE (3 mm diameter) as the working electrode, a platinum wire as the counter electrode and an Ag/AgCl as the reference electrode. The experimental conditions maintained for differential pulse voltammetric method were: pulse amplitude 50 mV; pulse width 30 ms and scan rate 20 mV s−1 while for square wave voltammetric method were: step potential 4.95 mV, amplitude 25.05 mV and scan rate 74.25 mV s−1.
2.4. Preparation of graphene oxide nanoribbons (GONRs) and electro-reduced GONRs
Pristine GONRs were synthesized by following the reported method.15 Briefly, 150 mg MWCNTs were suspended in 30 mL of conc. H2SO4 for 12 h. 750 mg KMnO4 (4.75 mM) was added to above mixture and stirred for 1 h at room temperature. Then, the reaction mixture was heated to 55–70 °C for 1 h. Further, the reaction mixture was quenched by pouring it in to ice-water containing a small amount of hydrogen peroxide. Finally, the solution was filtered off through polytetrafluoroethylene membrane filter paper and the separated solid was washed repeatedly with water and ethanol. The obtained solid was dried under IR lamp and it was designated as pristine GONRs. GONRs suspension was prepared in Millipore water by ultrasonication for about 1 h. Further, the film of GONRs was electrochemically reduced at GCE in the potential range of −1.7 to 0.6 V to obtain ErGONRs.18
2.5. Fabrication of GONRs/GCE and ErGONRs/GCE
GONRs/GCE was fabricated by the deposition of 6 μL GONRs dispersion on the surface of GCE and dried under IR lamp. It was then electrochemically reduced in phosphate buffer of pH 6 in the potential range of −1.7 to 0.6 V employing cyclic voltammetry (10 cycles) to produce ErGONRs/GCE (ESI, Fig. S1†).
3. Results and discussion
3.1. Characterization of GONRs and ErGONRs
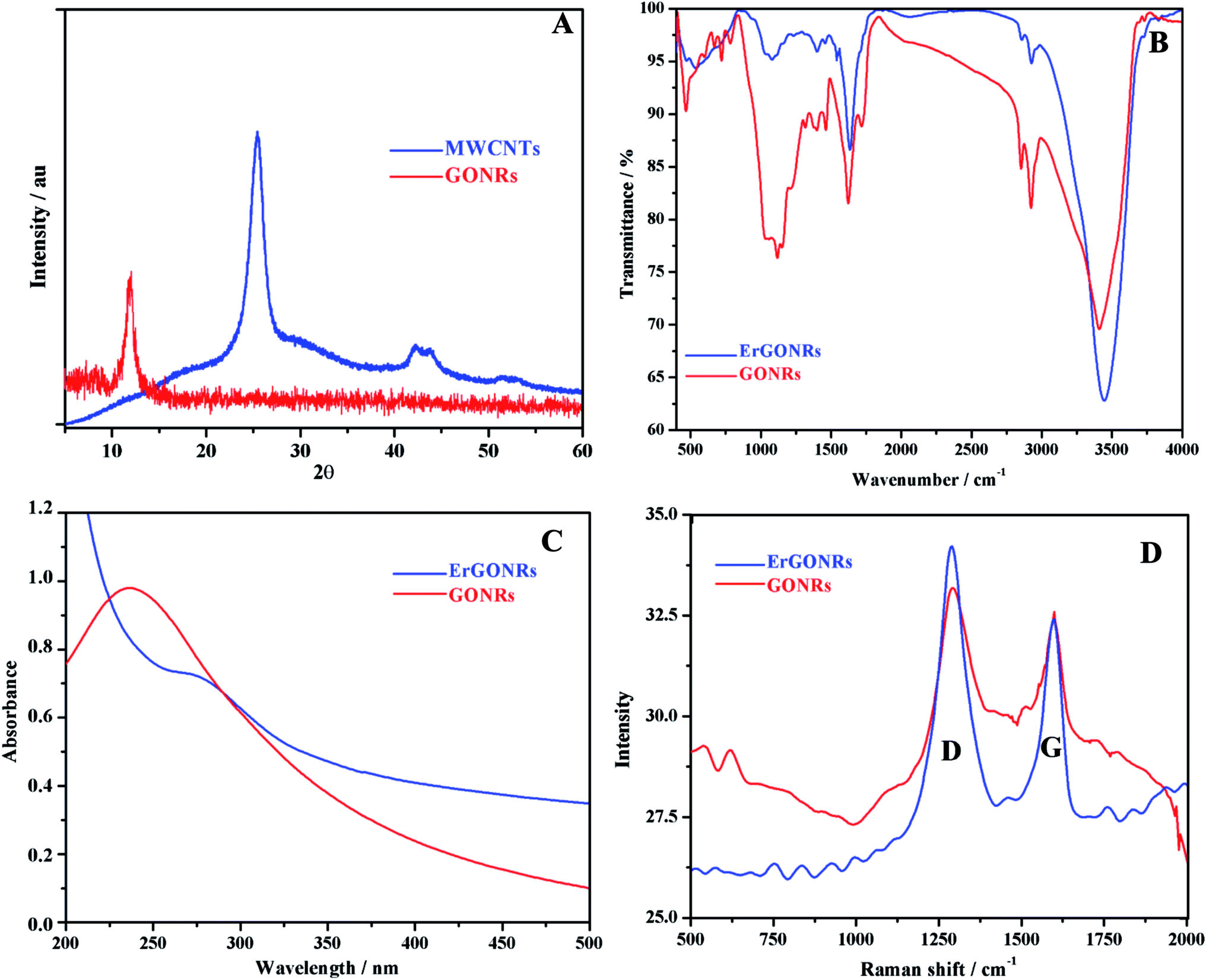
XRD studies. The XRD patterns of MWCNTs and GONRs are shown in Fig. 1A. Pristine MWCNTs exhibited characteristic diffraction peaks at 25.408° and 42.189° corresponding to (002) plane (d-spacing of 0.350 nm) and (101) plane (d-spacing of 0.214) respectively.61 Upon the oxidation of MWCNTs, the peak appeared at 10.06° corresponding to the (002) plane with an inter-planar distance of 0.878 nm. The larger d-spacing for GONRs compared to that of MWCNTs was attributed to the presence of intercalated oxygen containing functional groups and the formation of layered GONRs.
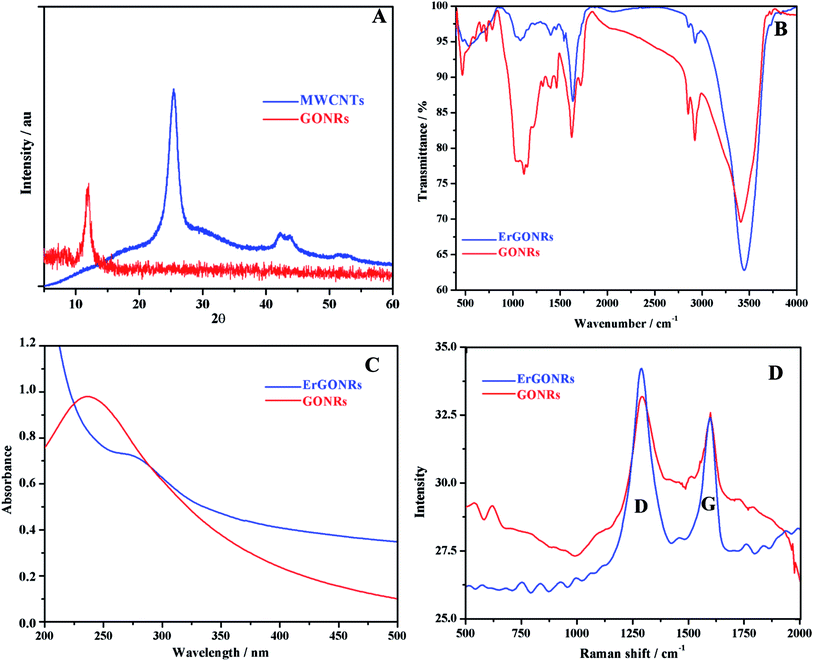 |
| | Fig. 1 (A) XRD patterns of MWCNTs (blue) and GONRs (red); (B) FTIR, (C) absorption and (D) Raman spectra of GONRs (red) and ErGONRs (blue). | |
FT-IR spectra. FT-IR spectra of GONRs and ErGONRs are shown in Fig. 1B. GONRs exhibited characteristic bands at 1715 cm−1 and 1030 cm−1 corresponding to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–OH stretching vibrations of the –COOH group respectively. The band at 1623 cm−1 was assigned to the bending vibration of absorbed water molecules and to contributions of sp2 characteristics whereas the band at 1214 cm−1 was attributed to the presence of epoxy groups. The bands noticed at 3413 cm−1 and 1400 cm−1 were due to the –OH stretching and deformation vibrations respectively. Upon electrochemical reduction of GONRs, the bands due to oxygen functionalities were observed to be almost disappeared indicating the restoration of sp2 hybridized carbon network. This facilitates the electron transfer kinetics when used as an electrode material.
O and C–OH stretching vibrations of the –COOH group respectively. The band at 1623 cm−1 was assigned to the bending vibration of absorbed water molecules and to contributions of sp2 characteristics whereas the band at 1214 cm−1 was attributed to the presence of epoxy groups. The bands noticed at 3413 cm−1 and 1400 cm−1 were due to the –OH stretching and deformation vibrations respectively. Upon electrochemical reduction of GONRs, the bands due to oxygen functionalities were observed to be almost disappeared indicating the restoration of sp2 hybridized carbon network. This facilitates the electron transfer kinetics when used as an electrode material.
Absorption studies. Absorption spectra of GONRs and ErGONRs are shown in Fig. 1C. GONRs exhibited an absorption band at ∼235 nm corresponding to π → π* transition of CC of GONRs network. Upon electrochemical reduction, the absorption band was shifted to ∼278 nm indicating that the π conjugations were extended or the small aromatic domains were restored.
Raman spectra. Raman spectra were recorded to evaluate the ordered and/or disordered structures of prepared GONRs and ErGONRs. The Raman shifts of GONRs and ErGONRs are shown in Fig. 1D. Prepared nanoribbons (GONRs and ErGONRs) exhibited D and G bands in the range of 1330–1592 cm−1, similar to that reported for graphene derivatives. The D band is related to the vibrational mode of the k-point phonon of A1g symmetry, whereas the G band is due to the E2g phonon of the sp2 hybridized carbon atoms.62 Further, the D/G ratio was found to be 0.957 and 1.158 for GONRs and ErGONRs respectively. The D/G ratio was increased upon electrochemical reduction of GONRs (i.e. the process of removal of intercalated oxygen-containing functional groups). This might be due to increase in the number of small aromatic domains responsible for the D band, but not necessarily their overall size, which is responsible for the G band or increased edge defects of narrow width of nanoribbons.63
SEM. The morphology and microstructures of prepared samples were investigated by FESEM. The micrographs of MWCNTs, GONRs and ErGONRs are shown in Fig. 2A–F. Carbon nanotubes were found to be agglomerated in pristine MWCNTs (Fig. 2A and B) with sizes ranging from 25–30 nm. Smooth and ribbon-like surface morphology was observed upon oxidative unzipping of MWCNTs (Fig. 2C and D). The width of such nanoribbons was found to be about 210 nm. This suggested that the MWCNTs were unzipped during the oxidation by KMnO4. Upon electrochemical reduction of GONRs, the surface morphology of nanoribbons became rougher and most of the edge planes were directed towards the surface (Fig. 2E and F). During electrochemical reduction process, first the carbonyl groups were reduced to form hydroxyl groups. The hydroxyl groups were further reduced by expulsion of oxygen in the form water molecules. Thus, the roughening of the substrate was due to the evolution of water molecules from the substrate. The evolved reduction products created various vacancies or defective sites that form rough surface upon electrochemical reduction of GONRs. These results revealed that the electron transfer proceeded at a faster rate at ErGONRs/GCE than that at GONRs/GCE.
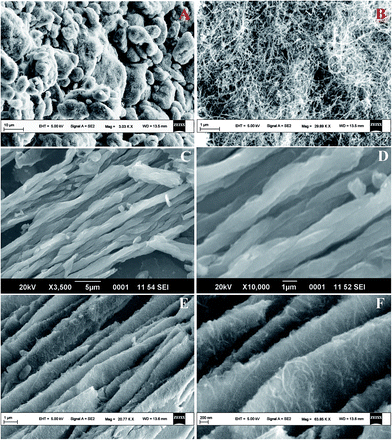 |
| | Fig. 2 SEM micrographs of MWCNTs (A and B), GONRs (C and D) and ErGONRs (E and F). | |
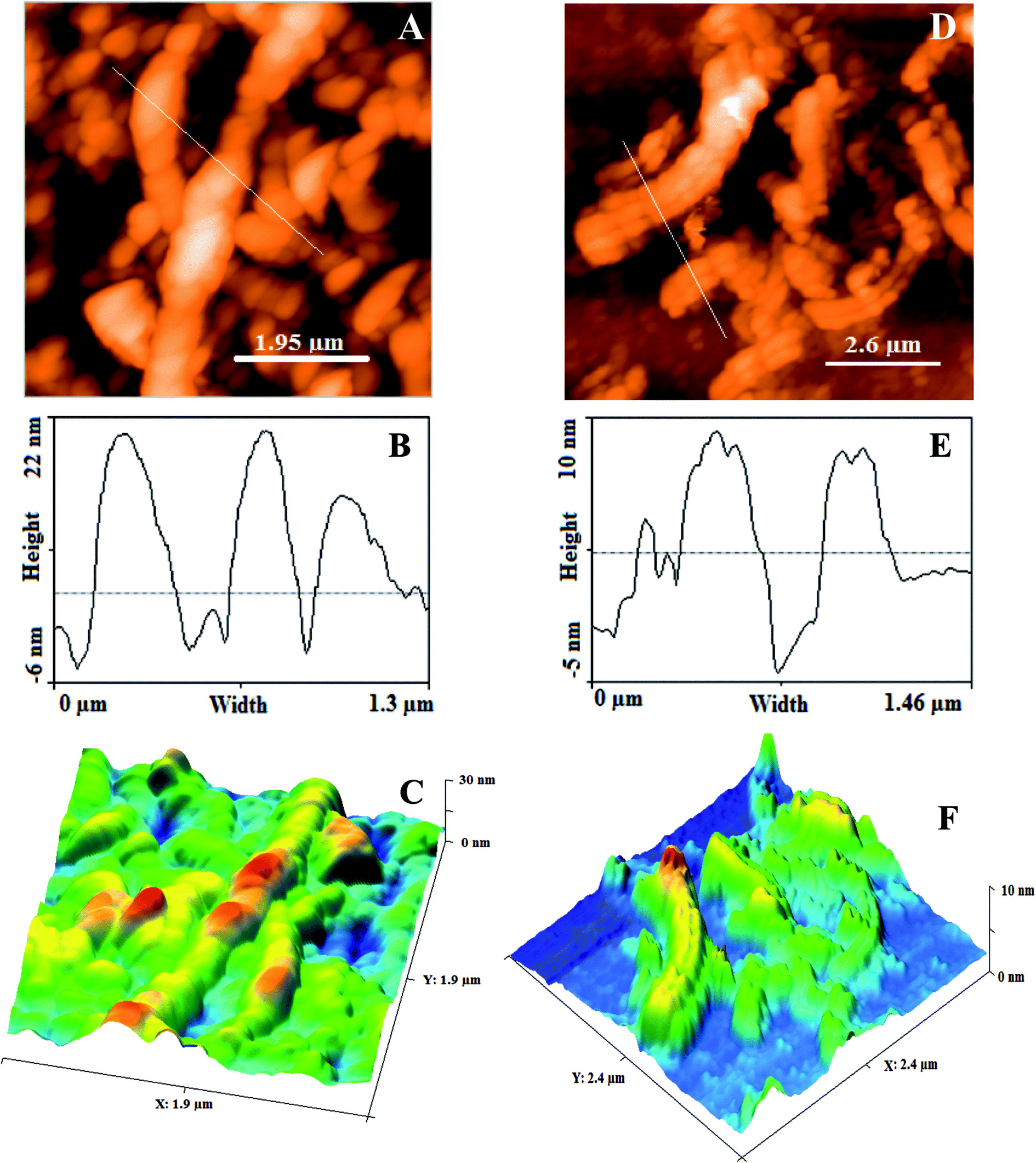
AFM. The structural features (surface roughness, length and width) of GONRs and ErGONRs were explored by AFM measurements. AFM, depth profile and 3D-AFM micrographs of GONRs and ErGONRs are shown in Fig. 3A–F. Upon oxidative unzipping, the length and width of MWCNTs were observed to be ∼2 μm and ∼225 nm respectively (Fig. 3A–C). This confirmed that the intercalation of oxygen functionalities into the CNTs network was successful. Upon electrochemical reduction of GONRs, the length and width was noticed to be ∼100 nm and ∼150 nm respectively (Fig. 3D–F). Upon electrochemical reduction, the width of GNRs was decreased by ∼75 nm, suggesting the reduction of oxygen functionalities at the edges. Surface roughness of ErGONRs (5.80 pm2) was found to be more than that observed for GONRs (3.81 pm2).
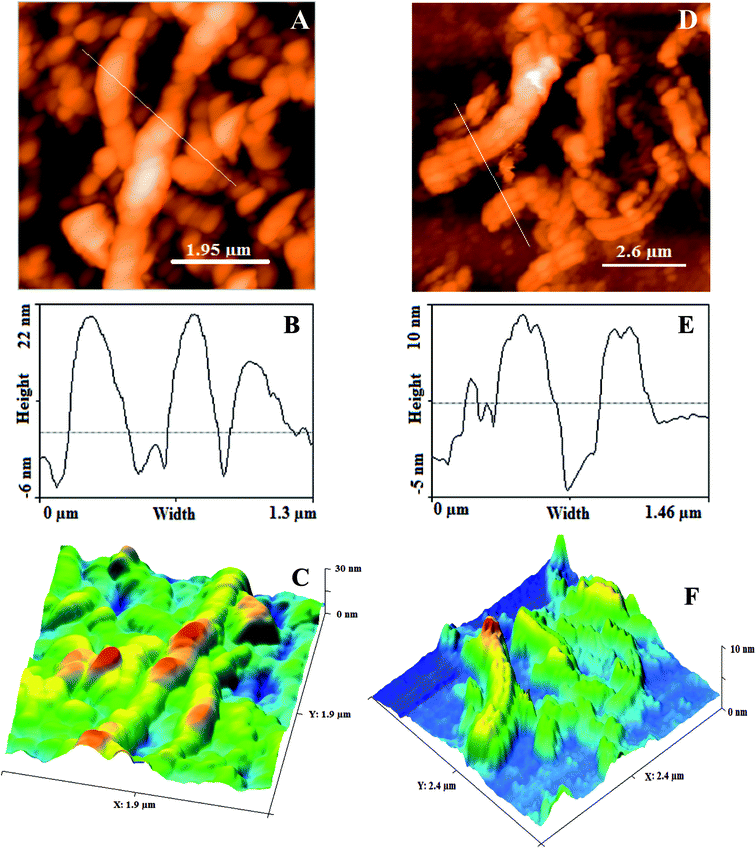 |
| | Fig. 3 AFM micrograph, height profile diagram and 3D AFM micrograph of GONRs (A–C) and ErGONRs (D–F). | |
3.2. Electrochemical characterization of GONRs/GCE and ErGONRs/GCE
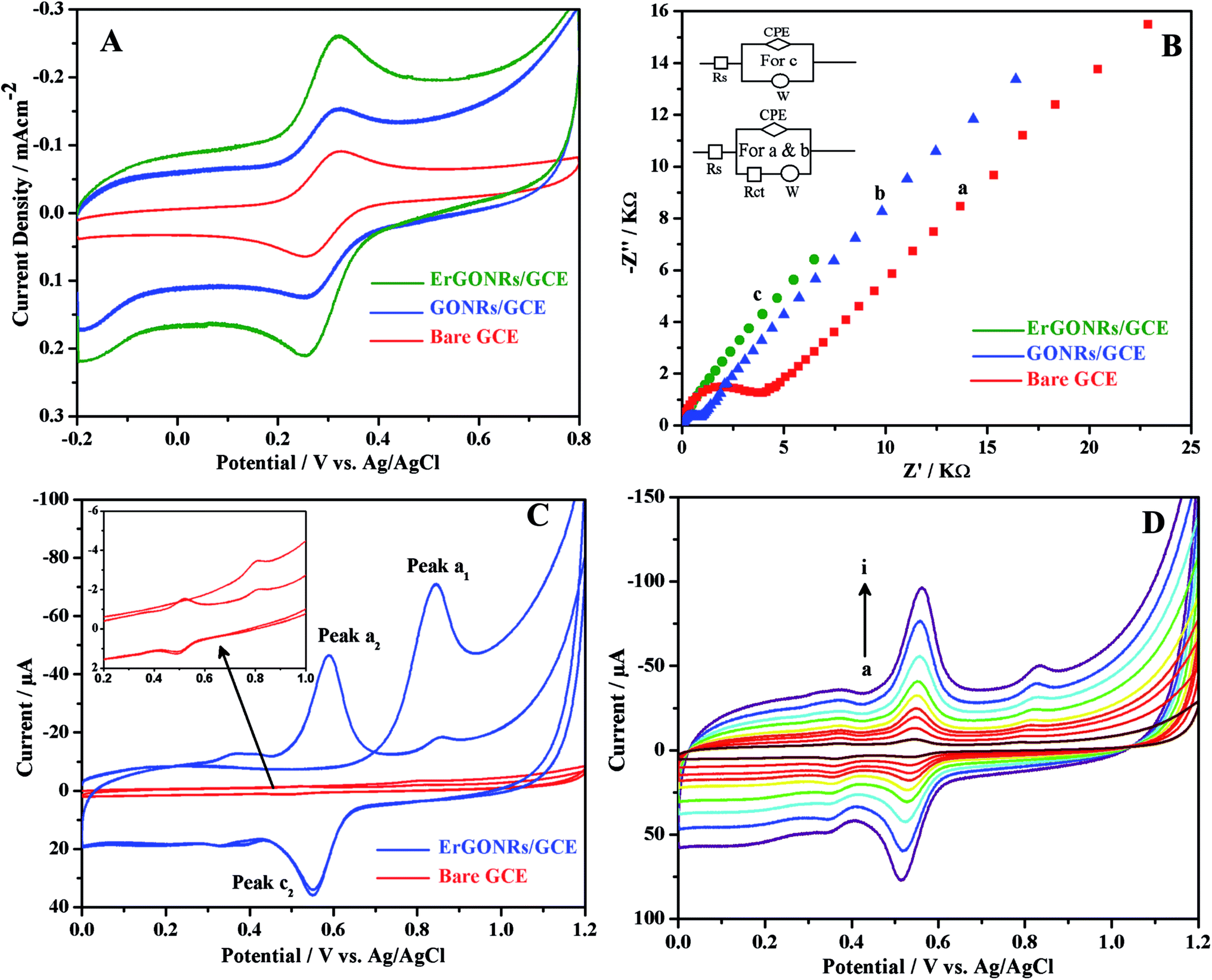
Cyclic voltammetric and electrochemical impedance spectroscopic techniques were used to characterize the stepwise fabrication process of the proposed electrochemical sensor. Cyclic voltammetric response of 1 mM potassium ferricyanide (redox probe) in 1 M KCl at bare GCE (curve a), GONRs/GCE (curve b) and ErGONRs/GCE (curve c) are shown in Fig. 4A. The probe exhibited higher redox peak current (∼47 times) at ErGONRs/GCE than that at GONRs/GCE. The lower redox response at GONRs/GCE was due to the electrostatic repulsion between oxygen-containing functional groups on GONRs network and ferricyanide anions. This repulsion restrained the diffusion of probe ions to the electrode surface. Moreover, lesser redox potential difference (ΔEp = 55 mV) was noticed at ErGONRs/GCE than that observed at GONRs/GCE (ΔEp = 63 mV) and bare GCE (ΔEp = 68 mV), indicating that the interface ErGONRs has outstanding charge transfer and catalytic performance. As expected, the response (increased Ip and decreased ΔEp) at ErGONRs/GCE was better than that at GONRs/GCE and bare GCE confirming the presence of several number of small aromatic domains/edge/basal planes in ErGONRs. The surface area of ErGONRs/GCE, GONRs/GCE and bare GCE was also calculated using Randles–Sevcik equation,64 Ip = 2.69 × 105n3/2AC0DR1/2v1/2 where Ip refers to the peak current, n is the number of electron transferred, A is the surface area of the electrode, DR is the diffusion coefficient, C0 is the concentration of K3[Fe(CN)6] and ν is the scan rate. For 1 mM K3[Fe(CN)6] in 0.1 M KCl electrolyte, n = 1 and DR = 7.6 × 10−6 cm s−1. From the slope of the plot of Ip vs. ν1/2, the electrochemically active surface area of ErGONRs/GCE, GONRs/GCE and bare GCE was calculated to be 0.62, 0.35 and 0.05 cm2 respectively. Hence, the ErGONRs/GCE was used as an electrochemical sensor.
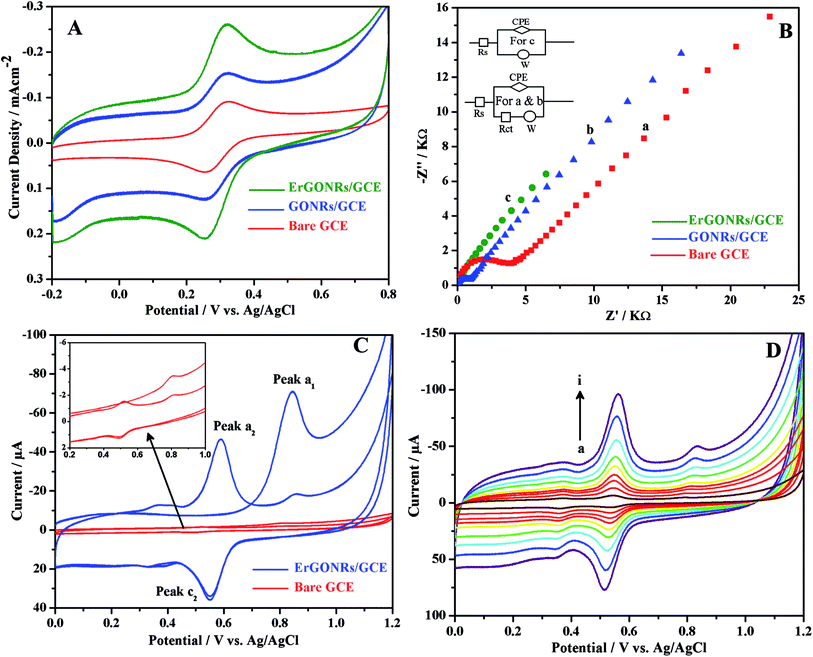 |
| | Fig. 4 (A) Cyclic voltammograms and (B) impedance spectra of 1 mM [Fe(CN)6]3−/4− at ErGONRs/GCE (green), GONRs/GCE (blue) and bare GCE (red) in 1 M KCl. (C) Cyclic voltammograms of 10 μM DIO at ErGONRs/GCE (blue) and bare GCE (red) in phosphate buffer of pH 3 and (D) cyclic voltammograms of 10 μM DIO at different scan rates (a → i: 20, 40, 60, 80, 100, 140, 180, 220 and 260 mV s−1, respectively) on ErGONRs/GCE in phosphate buffer of pH 3. | |
In the impedance spectrum, the semicircle diameter corresponds to the charge transfer resistance (Rct). This resistance controls the charge transfer kinetics of the redox probe (potassium ferricyanide) at the electrode interface. The Nyquist plots obtained at bare GCE (curve a), GONRs/GCE (curve b) and ErGONRs/GCE (curve c) in 1 M KCl solution containing 1 mM K3[Fe(CN)6] are shown in Fig. 4B. The charge transfer resistance, (Rct) of ErGONRs/GCE (Rct = 3.15 kΩ cm2) was found to be less than that observed for GONRs/GCE (Rct = 10.88 kΩ cm2) and bare GCE (Rct = 26.08 kΩ cm2), suggesting that the ErGONRs interface facilitated heterogeneous charge transfer rate. The corresponding equivalent circuits are shown in the inset of Fig. 4B. Thus, the results from electrochemical impedance measurements supported the results from voltammetric measurements. The slope of the diffusive part of the Nyquist plot was almost similar in GONRs/GCE and in ErGONRs/GCE. This indicated that the mass transfer of analyte to the substrate was not affected due to the reduction of GONRs. The removal of functional groups on electrochemical reduction of GONRs has improved the electrical conductivity which has resulted in the transport of charge or electrons through the modified electrode substrate.
3.3. Electrochemistry of DIO at ErGONRs/GCE
The proposed electrochemical sensor was utilized for investigating the detailed electrochemical characterization of DIO by employing cyclic voltammetric technique. Cyclic voltammograms of 10 μM DIO at ErGONRs/GCE and bare GCE in phosphate buffer of pH 3 are shown in Fig. 4C. DIO exhibited an oxidation peak (peak a1) at 0.845 V in the forward scan. Upon reversing the scan direction no reduction peak was observed corresponding to peak a1. A new reduction peak (peak c2) was noticed at 0.548 V and upon repeated cycling (in second cycle) a corresponding oxidation peak was observed (peak a2) at 0.587 V. Based on Testa–Reinmuth notation the proposed electrode reaction was believed to follow ECirrevE mechanism. Further, no reduction wave and corresponding oxidation wave was noticed upon reversing first cycle forward scan at 0.700 V (ESI, Fig. S2†). This confirmed that the DIO underwent irreversible electrooxidation in the first step and then chemically converted into electrochemically active product which was further involved in electrochemical redox reaction.
Redox wave potential of DIO was shifted to 62.2 mV per pH towards negative potential with increase in pH of the solution (from pH 3 to 6) indicating the involvement of equal number of protons and electrons in the electrode reaction. Irreversible oxidation peak was shifted to 32.5 mV per pH towards negative potential with increase in pH of the analyte solution (from pH 3 to 6) suggesting that one proton and two electrons have taken part in the electrode process.
Peak currents of all the peaks were observed to be linearly dependent on the scan rate in the range of 20–260 mV s−1 (Fig. 4D) suggesting that the electrode process was adsorption limited. The slope (∼0.85) of the logIp–logν plot further confirmed the adsorption controlled electrode process. Electrochemical kinetic parameters viz., number of electrons transferred (n), electron transfer coefficient (α) and rate of the reaction were evaluated by subjecting the scan rate results to Laviron equations shown below:65
| | |
Epa = E°′ + RT/αnF[ln(RTks)/(αnF) − lnν] (for irreversible system)
| (1) |
| |
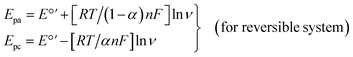 | (2) |
| | |
logks = αlog(1 − α) + (1 − α)logα − log(RT/nFν) − α(1 − α){(nFΔEp)/(2.3RT)}
| (3) |
where
E°′ is the formal potential,
n is the number of electrons transferred,
α is the electron transfer coefficient and
ks is the standard rate constant of the electrode reaction. For irreversible oxidation reaction, the values of
ks and
αn were deduced from the intercept and slope of the linear plot of
Epa vs. ln
ν, when the value of
E°′ was known. The value of
E°′ for DIO at ErGONRs/GCE was obtained from the intercept of
Epa vs. ν plot. Knowing the values of
E°′, the slope and intercept of
Epa vs. ln
ν plot (figure not shown), the values of
αn and
ks could be calculated and were found to be 1.09 and 1.54 s
−1 respectively. Since, for a totally irreversible electron transfer reaction,
α was assumed to be 0.5, the value of
n was calculated to be 2.19. This indicated the involvement of two electrons in the irreversible oxidation step. For redox reaction, the values of
α and
n were evaluated to be 0.53, 4.00 respectively by employing
eqn (2). By knowing the values of
α,
n, Δ
Ep(0.204/
n V) with the scan rate of 250 mV s
−1, standard rate constant (
ks) was evaluated using
eqn (3) and found to be 1.86 s
−1. From these results, we propose that two and four electrons were involved in the irreversible oxidation peak and redox peak, respectively.
3.4. Optimization of ErGONRs/GCE sensing performance
Sensing performance of ErGONRs/GCE was optimized by varying the amount of modifier (GONRs suspension), accumulation time and pH. DIO exhibited highest electrochemical response at ErGONRs/GCE in phosphate buffer of pH 3 with an accumulation time of 210 s (ESI, Fig. S3†) and 6 μL GONRs suspension (ESI, Fig. S4†). Hence, these parameters were fixed for further studies.
4. Analytical applications of ErGONRs/GCE
4.1. Construction of analytical curve
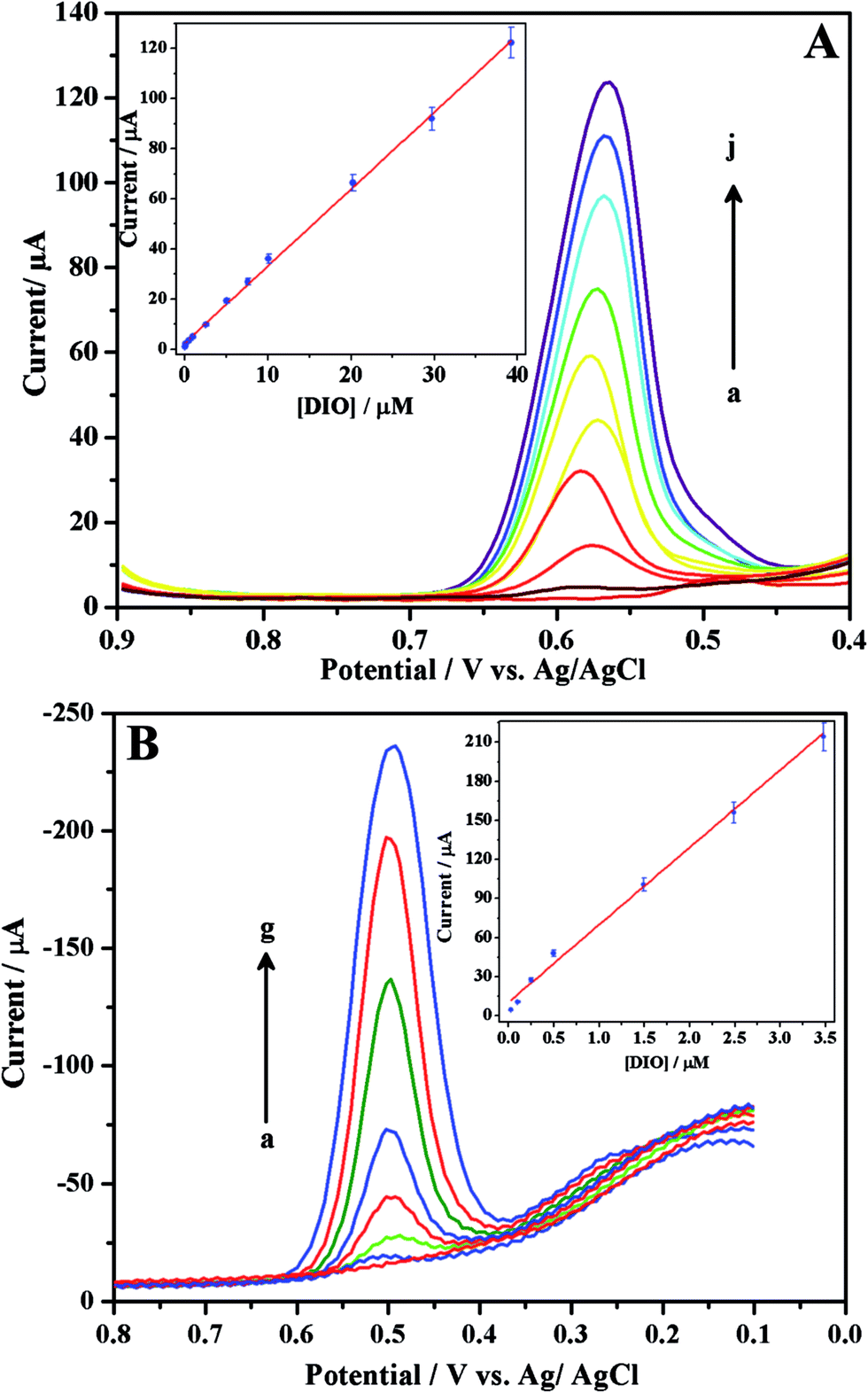
With the aim of proposing sensitive electroanalytical methods for the assay of DIO, we have developed SWV and DPV methods. Among these two methods, SWV method is more sensitive compared to that of DPV as evident from their low detection limits. In order to construct the calibration curves, differential pulse (Fig. 5A) and square wave (Fig. 5B) voltammograms were recorded for increasing concentrations of DIO. Linearity was observed between the peak current and concentration of DIO in the range of 51.01 nM–39.21 μM and 25 nM–3.48 μM for differential pulse (inset of Fig. 5A) and square wave (inset of Fig. 5B) voltammetric methods respectively. The corresponding regression equations are shown below:
| Ip/μA = 3.06[DIO] + 2.55; r = 0.998 (for DPV method) |
| Ip/μA = 61.4[DIO] + 12.1; r = 0.992 (for SWV method) |
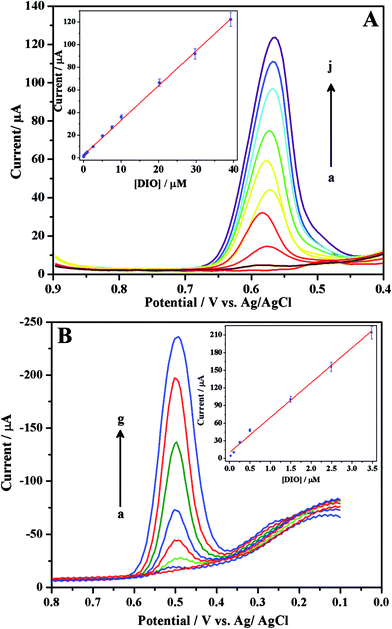 |
| | Fig. 5 Differential (A) and square wave (B) voltammograms for increasing concentrations ((A) (a → j) 0.0510, 0.102, 0.510, 1.20, 2.54, 5.07, 10.1, 20.2, 29.7 and 39.21 μM, respectively and (B) (a → g) 0.0250, 0.0995, 0.249, 0.498, 1.49, 2.49 and 3.48 μM, respectively) of DIO in phosphate buffer of pH 3. Insets show respective linear plots. | |
The values of limit of detection (LOD) and limit of quantification (LOQ) were evaluated and the corresponding results are shown in Table 1. The values of RSD less than 2.81% confirmed that the developed methods were sufficiently precise for the sensing of DIO. Further, the analytical characteristics (analytical range and limit of detection) of the proposed method were compared with those of reported methods (Table 2).
Table 1 Characteristics of the calibration plots for DIO
| |
DPVa |
SWVb |
| Differential pulse voltammetric method. Square wave voltammetric method. For 1 μM DIO. |
| Linearity range, μM |
0.051–39.21 |
0.025–3.48 |
| LOD, μM |
0.015 |
0.011 |
| LOQ, μM |
0.049 |
0.040 |
| Inter-day assay RSDc (%) |
2.81 |
2.34 |
| Intra-day assay RSDc (%) |
2.56 |
2.53 |
Table 2 Comparison of analytical range and detection limits of the proposed method with those of reported methods
| Method |
Linearity range (μg mL−1) |
LOD (μg mL−1) |
References |
| Spectrophotometric |
| Method-1 |
0.3–35 |
0.1 |
33 |
| Method-2 |
2–140 |
0.05 |
34 |
|
| Chromatographic |
| HPLC |
0.08–0.15 |
— |
35 |
| HPTLC (per spot) |
0.10–3.0 |
0.1 |
39 |
|
| Electrochemical |
| AdSV (GCE) |
0.50–165.5 |
0.20 |
42 |
| SWV (CPE) |
— |
1.62 |
43 |
| DPV (ErGONRs/GCE) |
0.03–23.83 |
0.03 |
Proposed work |
| SWV (ErGONRs/GCE) |
0.015–1.85 |
0.02 |
4.2. Assay of DIO in pharmaceutical formulations
The practical utility of the proposed sensor, ErGONRs/GCE was established by determining DIO in pharmaceutical formulations. The corresponding results of the analysis are tabulated in Table 3 and found to be satisfactory. Further, recovery studies were performed to check the possible interferences from the common excipients and additives. For this, known amounts of pure DIO were added to the previously analyzed formulation of DIO and analyzed. High percentage of recovery indicated that the commonly encountered excipients and additives did not interfere in the assay by the proposed method.
Table 3 Results of analysis of DIO in pharmaceutical formulations
| |
VENUSMINa |
| Martin and Harris Laboratories, LTD., India. Average of six determinations. For 1 μM DIO. |
| Labelled amount, mg |
150.0 |
| Amount found, mg |
148.93 |
| Recoveryb, % |
99.28 |
| RSDc, % |
0.42 |
| Pure DIO added to tablet solution, mg |
25.00 |
| Amount found, mg |
24.50 |
| Recoveryb, % |
98.0 |
| RSDc, % |
2.11 |
5. Binding characteristics of DIO–HSA at ErGONRs/GCE
5.1. Interaction of DIO with HSA by CV and DPV
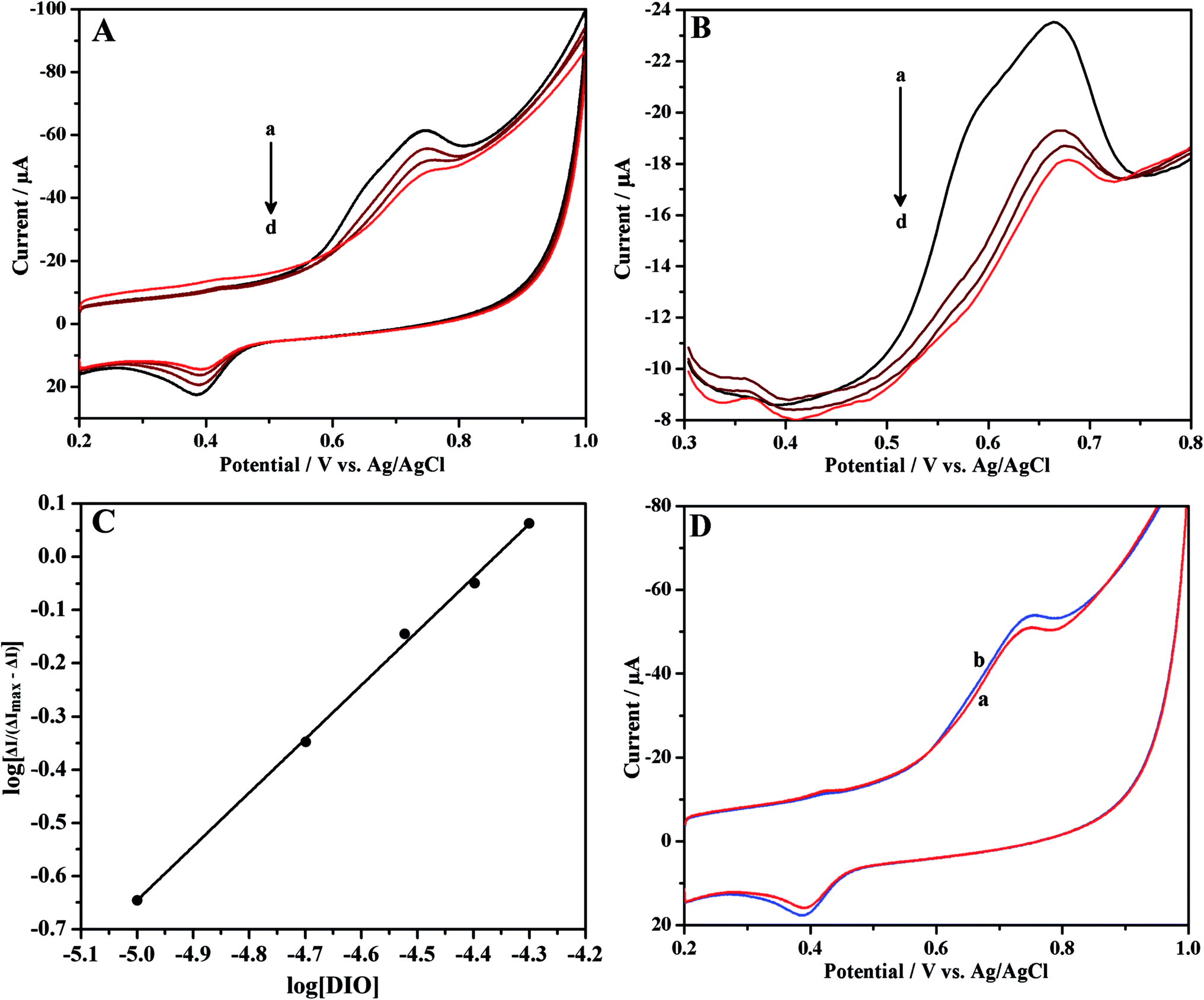
Cyclic and differential pulse voltammetric techniques were used to investigate the interaction between DIO and HSA at ErGONRs/GCE in phosphate buffer of physiological pH. Cyclic and differential pulse voltammograms of DIO at ErGONRs/GCE in the presence of increasing amounts of HSA are shown in Fig. 6A and B. Significant decrease in the oxidation peak (a1) current and positive shift in the peak potential of DIO was observed upon the addition of HSA. Further, no new peak was observed in the presence of HSA. The drop in oxidation peak current was noticed to be significant up to 4 min. After 4 min the peak current remained constant. Therefore, the interaction time of 4 min was maintained for each measurement. The drop in oxidation peak current was due to one of the two reasons viz., (i) the formation of an electrochemically inactive supramolecular DIO–HSA complex which decreased the concentration of free DIO or (ii) the addition of HSA might alter the electrochemical kinetics of DIO.66–68 In order to confirm this, the electrochemical kinetic parameters of DIO in the presence and absence of HSA were evaluated. The values of ks and αn were calculated to be 1.53 s−1 and 1.11 and, 1.51 s−1 and 1.10 in the absence and presence of HSA respectively (Table 4). These results did not reveal any significant changes in electrochemical kinetic parameters in the presence and absence of HSA. Hence, we propose that the decreased peak current of DIO upon the addition of HSA was due to the formation of an electrochemically inactive complex, DIO–HSA.
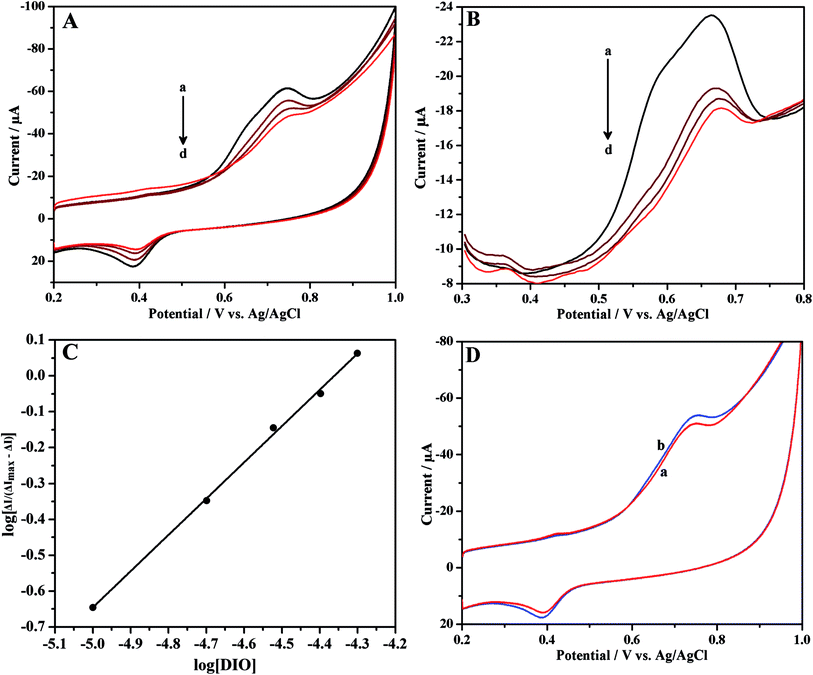 |
| | Fig. 6 (A) Cyclic and (B) differential pulse voltammograms of 10 μM DIO in the presence of increasing concentrations of HSA (a → d: 0, 5, 10 and 15 μM, respectively), at ErGONRs/GCE in phosphate buffer of pH 7. (C) Linear plot of log[ΔI/(ΔImax − ΔI)] versus log[DIO]. (D) Cyclic voltammograms of 10 μM DIO at ErGONRs/GCE (a) and HSA modified ErGONRs/GCE in phosphate buffer of pH 7. | |
Table 4 Electrochemical parameters for DIO in the presence and absence of HSA
| |
E°′ (V) |
nα |
ks (s−1) |
m |
K, 104 (mol−1) |
| Free DIO |
0.698 |
1.11 |
1.53 |
— |
— |
| DIO–HSA |
0.700 |
1.10 |
1.51 |
1.01 |
2.58 |
5.2. Investigation of binding parameters for DIO–HSA system
According to reports available in literature,69–71 it is assumed that DIO interacted with HSA to produce only a single complex, HSA–mDIO, as shown below:
where m is the binding ratio.
The equilibrium constant, βS is deduced as follows:
| | |
βS = [HSA–mDIO]/{[HSA][DIO]m}
| (4) |
because
| CHSA = [HSA] + [HSA–mDIO] |
According to the Ilkovič equation of an quasireversible electrode process,
| | |
[HSA] + [HSA–mDIO] = CHSA
| (7) |
| | |
ΔImax − ΔI = k(CHSA − [HSA–mDIO])
| (8) |
From eqn (4), (5) and (9) we get
| | |
(1/ΔI) = (1/ΔImax) + {[1/(βSΔImax)] × (1/[DIO]m)}
| (10) |
or
| | |
log[ΔI/(ΔImax − ΔI)] = logβS + mlog[DIO]
| (11) |
where Δ
I is the difference in peak current of DIO in the presence and absence of HSA and Δ
Imax is the peak current of DIO at extremely higher concentration compared to that of HSA.
CHSA, [HSA] and [HSA–
mDIO] correspond to the total, free and bound concentration of HSA in the solution, respectively.
The plot of log[ΔI/(ΔImax − ΔI)] versus log[DIO] (Fig. 6C) shows linearity and slope equal to m, if the interaction of HSA with DIO yields a single complex. The values of m and βS were deduced from the slope and intercept and were found to be 1.01 and 2.58 × 104 M−1, respectively (Table 4). This indicated the formation of a stable 1:1 complex of HSA–DIO. The values of binding constant (βS) and stoichiometry of the HSA–DIO complex are in agreement with reported values (obtained by spectroscopic methods).71,72
5.3. Interaction of DIO with HSA–ErGONRs/GCE
The immobilization of HSA onto an electrode surface is in many ways the crucial aspect to develop HSA biosensors for monitoring the drug since it dictates the accessibility of HSA to drug in solution and hence can influence the affinity of drug binding.73,74 In view of this, we have carried out the interaction studies at HSA–ErGONRs/GCE that was prepared by immobilization technique. Cyclic voltammograms of 10 μM DIO at ErGONRs and HSA–ErGONRs/GCE in the potential range of 0.2–1 V are shown in Fig. 6D.
Oxidation peak (peak a1) potential was shifted towards positive potential (∼10 mV) at HSA–ErGONRs/GCE when compared to that at ErGONRs/GCE. The positive shift in the oxidation potential at HSA–ErGONRs/GCE suggested the interaction between DIO and HSA. Heli et al.73 have attributed the positive and negative shifts observed at HSA/GCE electrode to hydrophobic and non-hydrophobic (electrostatic and/or covalent) interactions, respectively. The positive shift observed in the present study indicated the interaction between DIO and HSA to be hydrophobic forces.
Conclusions
An electrochemical sensor was fabricated based on eco-friendly reduction of graphene oxide nanoribbons for sensing applications. Graphene oxide nanoribbons were prepared by oxidative unzipping of multiwalled carbon nanotubes. These oxidized nanoribbons were electrochemically reduced to regain small aromatic domains for electrosensing applications. Electrochemical sensor was constructed by drop casting of graphene oxide nanoribbon suspension on GCE followed by electrochemical reduction. Application of the proposed electrochemical sensor was also demonstrated by exploring the binding mechanism of DIO–HSA system.
Acknowledgements
The authors are grateful to BRNS, BARC, Mumbai, for the financial support (no. 2012/37C/8/BRNS/637 dated 28-05-2012). One of the authors (Nagappa L. Teradal) acknowledges DST, New Delhi for the award of INSPIRE Fellowship (IF110339).
References
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- H. B. Heersche, P. Jarillo-Herrero, J. B. Oostinga, L. M. K. Vandersypen and A. F. Morpurgo, Nature, 2007, 446, 56 CrossRef CAS PubMed.
- V. M. Tsefrikas and L. T. Scott, Chem. Rev., 2006, 106, 4868 CrossRef CAS PubMed.
- M. D. Watson, A. Fechtenkotter and K. Mullen, Chem. Rev., 2001, 101, 1267 CrossRef CAS PubMed.
- O. Vostrowsky and A. Hirsch, Chem. Rev., 2006, 106, 5191 CrossRef CAS PubMed.
- T. J. J. Mueller and U. H. F. Bunz, Functional Organic Materials, Wiley-VCH, Weinheim, 2007 Search PubMed.
- Y. Zhang, Y. W. Tan, H. L. Stromier and K. Kim, Nature, 2005, 438, 201 CrossRef CAS PubMed.
- C. Berger, Z. Song, X. Li, X. Wu, N. Brown, C. Nafud, D. Mayo, T. Li, A. N. Marchenkov, E. H. Conrad, P. N. First and W. A. de Heer, Science, 2006, 312, 1191 CrossRef CAS PubMed.
- T. Ohta, A. Bostwick, T. Seyller, K. Horn and E. Rotenberg, Science, 2006, 313, 951 CrossRef CAS PubMed.
- X. Wang, X. Li, L. Zhang, Y. Yoon, P. K. Weber, H. Wang, J. Guo and H. Dai, Science, 2009, 324, 76 Search PubMed.
- L. Jiao, L. Zhang, X. Wang, G. Diankov and H. Dai, Nature, 2009, 458, 877 CrossRef CAS PubMed.
- L. Xie, H. Wang, C. Jin, X. Wang, L. Jiao, K. Suenaga and H. Dai, J. Am. Chem. Soc., 2011, 133, 10394 CrossRef CAS PubMed.
- L. Jiao, X. Wang, G. Diankov, H. Wang and H. Dai, Nat. Nanotechnol., 2010, 5, 321 CrossRef CAS PubMed.
- Z. Zhang, Z. Sun, J. Yao, D. V. Kosynkin and J. M. Tour, J. Am. Chem. Soc., 2009, 131, 13460 CrossRef CAS PubMed.
- D. V. Kosynkin, A. L. Higginbotham, A. Sinitskii, J. R. Lomeda, A. Dimiev, B. K. Price and J. M. Tour, Nature, 2009, 458, 872 CrossRef CAS PubMed.
- A. L. Elias, A. R. Botello-Mendez, D. Meneses-Rodriguez, V. J. Gonzalez, D. Ramirez-Gonzalez, L. Ci, E. Munoz-Sandoval, P. M. Ajayan, H. Terrones and M. Terrones, Nano Lett., 2010, 10, 366 CrossRef CAS PubMed.
- A. G. Cano-Marquez, F. J. Rodriguez-Macias, J. Campos-Delgado, C. G. Espinosa-Gonzalez, F. Tristan-Lopez, D. Ramirez-Gonzalez, D. A. Cullen, D. J. Smith, M. Terrones and Y. I. Vega-Cantu, Nano Lett., 2009, 9, 1527 CrossRef CAS PubMed.
- C. E. Chng, M. Pumera and A. Bonanni, Electrochem. Commun., 2014, 46, 137 CrossRef CAS PubMed.
- C. H. A. Wong and M. Pumera, J. Mater. Chem. C, 2014, 2, 856 RSC.
- C. K. Chua, Z. Sofer and M. Pumera, Chem.–Asian J., 2012, 7, 2367 CrossRef CAS PubMed.
- D. B. Shinde, J. Debgupta, A. Kushwaha, M. Aslam and V. K. Pillai, J. Am. Chem. Soc., 2011, 133, 4168 CrossRef CAS PubMed.
- H. L. Guo, X. F. Wang, Q. Y. Qian, F. B. Wang and X. H. Xia, ACS Nano, 2009, 3, 2653 CrossRef CAS PubMed.
- A. M. Browning, U. K. Walle and T. Walle, J. Pharm. Pharmacol., 2005, 57, 1037 CrossRef CAS PubMed.
- C. M. Conesa, V. V. Ortega, M. J. Y. Gascón, M. A. Baños, M. C. Jordana, O. Benavente-García and J. Castillo, J. Agric. Food Chem., 2005, 53, 6791 CrossRef PubMed.
- C. Martínez, V. Vicente, J. Yáñez, M. Alcaraz, M. T. Castells, M. Canteras, O. G. Benavente-García and J. Castillo, Histol. Histopathol., 2005, 20, 1121 Search PubMed.
- S. Zhang, C. Qin and S. H. Safe, Environ. Health Perspect., 2003, 111, 877 CrossRef.
- Q. Zheng, Y. Hirose and N. Yoshimi, J. Cancer Res. Clin. Oncol., 2002, 128, 539 CrossRef CAS PubMed.
- K. A. Lyseng-Williamson and C. M. Perry, Drugs, 2003, 63, 71 CrossRef CAS PubMed.
- G. Jantet, Angiology, 2002, 53, 245 CrossRef CAS PubMed.
- A. A. Ramelet, Angiology, 2001, 52, S49 CrossRef PubMed.
- J. J. Bergan, G. W. Schmid-Schonbein and S. Takase, Angiology, 2001, 52, 43 CrossRef PubMed.
- D. MacKay, Altern. Med. Rev., 2001, 6, 126 CAS.
- Z. Moldovan, A. A. Bunaciu, M. A. Al-Omar and H. Y. Aboul-Enein, Open Chem. Biomed. Methods J., 2010, 3, 123 CrossRef CAS.
- Z. Moldovan and H. Y. Aboul-Enein, Instrum. Sci. Technol., 2011, 39, 135 CrossRef CAS PubMed.
- I. Saeidi, M. R. Hadjmohammadi, M. Peyrovi, M. Iranshahi, B. Barfi, A. B. Babaei and A. M. Dust, J. Pharm. Biomed. Anal., 2011, 56, 419 CrossRef CAS PubMed.
- M. A. Campanero, M. Escolar, G. Perez, E. Garcia-Quetglas, B. Sadaba and J. R. Azanza, J. Pharm. Biomed. Anal., 2010, 51, 875 CrossRef CAS PubMed.
- F. I. Kanaze, C. Gabrieli, E. Kokkalou, M. Georgarakis and I. Niopas, J. Pharm. Biomed. Anal., 2003, 33, 243 CrossRef CAS.
- A. M. El-Shafae and M. M. El-Domiaty, J. Pharm. Biomed. Anal., 2001, 26, 539 CrossRef CAS.
- E. Shawky, J. Planar Chromatogr.–Mod. TLC, 2012, 25, 138 CrossRef CAS.
- I. Adouani, M. Du and T. J. Hang, Chromatographia, 2013, 76, 499 CAS.
- V. Adam, R. Mikelova, J. Hubalek, P. Hanustiak, M. Beklova, P. Hodek, A. Horna, L. Trnkova, M. Stiborova, L. Zeman and R. Kizek, Sensors, 2007, 7, 2402 CrossRef CAS PubMed.
- M. S. El-Shahawi, A. S. Bashammakh and T. El-Mogy, Anal. Sci., 2006, 22, 1351 CrossRef CAS.
- P. Hanuštiak, R. Mikelová, D. Potěšil, P. Hodek, M. Stiborová and R. Kizek, Biomed. Pap., 2005, 149, 44 Search PubMed.
- J. Wei, F. Jin, Q. Wu, Y. Jiang, D. Gao and H. Liu, Talanta, 2014, 126, 116 CrossRef CAS PubMed.
- V. Sinisi, C. Forzato, N. Cefarin, L. Navarini and F. Berti, Food Chem., 2015, 168, 332 CrossRef CAS PubMed.
- F. S. Mohseni-Shahri, M. R. Housaindokht, M. R. Bozorgmehr and A. A. Moosavi-Movahedi, J. Lumin., 2014, 154, 229 CrossRef CAS PubMed.
- D. Xiao, L. Zhang, Q. Wang, X. Lin, J. Sun and H. Li, J. Lumin., 2014, 146, 218 CrossRef CAS PubMed.
- L. Tang, H. Zuo and S. Li, J. Lumin., 2014, 153, 54 CrossRef CAS PubMed.
- Y. Y. Yue, X. G. Chen, J. Qin and X. J. Yao, J. Pharm. Biomed. Anal., 2009, 49, 756 CrossRef PubMed.
- D. C. Carter and J. X. Ho, Adv. Protein Chem., 1994, 45, 153 CAS.
- J. Seetharamappa, B. Sandhya, A. H. Hegde, N. L. Teradal and S. N. Prashanth, Curr. Drug Metab., 2013, 14, 456 CrossRef.
- A. H. Hegde, B. Sandhya and J. Seetharamappa, Mol. Biol. Rep., 2011, 38, 4921 CrossRef CAS PubMed.
- S. Chaudhuri, B. Sengupta, J. Taylor, B. P. Pahari and P. K. Sengupta, Curr. Drug Metab., 2013, 14, 491 CrossRef CAS.
- X. Zhang, L. Li, Z. Xu, Z. Liang, J. Su, J. Huang and B. Li, PLoS One, 2013, 8, e59106 CAS.
- G. Zhang, L. Wang and J. Pan, J. Agric. Food Chem., 2012, 60, 2721 CrossRef CAS PubMed.
- C. Z. Lin, M. Hu, A. Z. Wu and C. C. Zhu, J. Pharm. Anal., 2014, 4, 392 CrossRef PubMed.
- J. Dai, T. Zou, L. Wang, Y. Zhang and Y. Liu, Luminescence, 2014, 29, 1154 CrossRef CAS PubMed.
- C. Z. Lin, M. Hu, A. Z. Wu and C. C. Zhu, J. Pharm. Anal., 2014, 4, 392 CrossRef PubMed.
- D. K. F. Meijer and P. Van der sluijs, Pharm. Res., 1989, 6, 105 CrossRef CAS.
- S. Sugio, A. Kashima, S. Mochizuki, M. Noda and K. Kobayashi, Protein Eng., 1999, 12, 439 CrossRef CAS PubMed.
- K. Ding and G. Yang, Mater. Chem. Phys., 2010, 23, 498 CrossRef PubMed.
- H. Kim, D. H. Seo, S. W. Kim, J. Kim and K. Kang, Carbon, 2011, 49, 326 CrossRef CAS PubMed.
- A. L. Higginbotham, D. V. Kosynkin, A. Sinitskii, Z. Sun and J. M. Tour, ACS Nano, 2010, 4, 2059 CrossRef CAS PubMed.
- B. Rezaei and S. Damiri, Sens. Actuators, B, 2008, 134, 324 CrossRef CAS PubMed.
-
(a) E. Laviron, J. Electroanal. Chem., 1979, 101, 19 CrossRef CAS;
(b) E. Laviron and L. J. Roullier, Electroanal. Chem., 1980, 115, 65 CrossRef CAS.
- W. Sun, K. Jiao, X. L. Wang and L. D. Lu, J. Electroanal. Chem., 2005, 578, 37 CrossRef CAS PubMed.
- Z. W. Zhu and N. Q. Li, Microchem. J., 1998, 59, 294 CrossRef CAS.
- H. M. Zhang, Z. W. Zhu and N. Q. Li, Fresenius. J. Anal. Chem., 1999, 363, 408 CrossRef CAS.
- W. Suna, J. Han, K. Jiao and L. Lu, Bioelectrochemistry, 2006, 68, 60 CrossRef PubMed.
- S. S. Kalanur, J. Seetharamappa and U. Katrahalli, Colloids Surf., B, 2010, 75, 75 CrossRef CAS PubMed.
-
(a) L. Fun, X. Liu, Q. Zhou, J. Zhang, J. Dong and J. Wang, J. Lumin., 2014, 149, 208 CrossRef PubMed;
(b) D. Barreca, G. Laganà, G. Bruno, S. Magazù and E. Bellocco, Biochimie, 2013, 95, 2042 CrossRef CAS PubMed.
- C. Dufour and O. Dangles, Biochim. Biophys. Acta, 2005, 1721, 164 CrossRef CAS PubMed.
- H. Heli, N. Sattarahmady, A. Jabbari, A. A. Moosavi-Movahedi, G. H. Hakimelahi and F. Y. Tsai, J. Electroanal. Chem., 2007, 610, 67 CrossRef CAS PubMed.
- V. Brabec, Electrochim. Acta, 2000, 45, 2929 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra07497e |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here.