
Coordination versatility of phosphine derivatives of fluoroquinolones. New CuI and CuII complexes and their interactions with DNA†
A. Bykowska,
R. Starosta,
J. Jezierska and
M. Jeżowska-Bojczuk*
Faculty of Chemistry, University of Wrocław, ul. F. Joliot-Curie 14, 50-383 Wrocław, Poland. E-mail: malgorzata.jezowska-bojczuk@chem.uni.wroc.pl; Tel: +48 71 3757281
First published on 14th September 2015
Abstract
In this paper, new copper(I) and copper(II) complexes with phosphine derivatives of two fluoroquinolones (ciprofloxacin and norfloxacin) are presented. The synthesized compounds ([CuI-PCp], [CuI-PNr], [OPCp-CuII]+ and [OPNr-CuII]+) were characterized by elemental analysis and MS as well as by the NMR, EPR and IR spectroscopies. X-ray techniques were used to determine the crystal and molecular structures of [CuI-PCp]·CH2Cl2·CH3CN and OPCp-CuII]NO3·3H2O. For all the studied compounds, the ability to interact with DNA was determined using three different methods. The results of gel electrophoresis revealed that in the presence or absence of H2O2, the copper(I) complexes caused only single-stranded cleavage of the sugar–phosphate backbone of DNA. In turn, the copper(II) complexes damaged the plasmid exclusively in the presence of the oxidant. The addition of H2O2 caused distinct changes in the plasmid structure, resulting in a complete disappearance of its native form. Forms II and III arising from single- and double-strand cleavage were detected. Studies of the interactions with calf thymus DNA in the presence of ethidium bromide (EB) showed that the tested complexes and phosphines interact with DNA in a partial intercalation mode, contrary to unmodified antibiotic and oxide derivatives, which do not displace EB from the system. Molecular docking (AutoDock Vina program) was performed using the synthetic double-stranded hexadecanucleotide (sequence: ATATCGCGATATCGCG). Data analysis showed that a majority of the compounds preferably bound to the minor or major grooves, however most of them were also able to intercalate with the DNA double helix.
Introduction
The discovery of the anticancer properties of cisplatin in the mid-twentieth century resulted in intensive development of medicinal and bioinorganic chemistry. Metal complexes as potential therapeutics are very interesting compounds due to (i) the accessible redox states of metal ions, (ii) wide range of coordination numbers, (iii) interesting kinetic and thermodynamic properties.1–3 Recently much research attention has been paid to complexes with biologically active molecules, for example drugs, which are donor-rich compounds efficiently binding metal ions. Numerous literature reports state that coordination compounds often have better biological properties than uncoordinated drugs. Formation of a complex frequently changes the solubility, bioavailability and the mechanism of action of the parent molecule. The consequence of these changes can be better biological properties of the complex or its broader spectrum of activity in comparison to the free drug. In this context it is essential to recognize the potential molecular targets in the cell, by determining the ability of studied complexes to their interactions with the macromolecules present in living organisms (DNA, RNA, proteins).4–9Metal ions complexes with fluoroquinolones, aminoglycosides, β-lactams, tetracyclines and peptide antibiotics interact with DNA in a different way than uncoordinasted drugs. They are able to damage plasmid DNA generating single and double strand breaks in a sugar–phosphate backbone. Complexes can also bind to minor either major groove of DNA molecule or act as intercalating agent penetrating into the complementary nitrogen base pairs of the DNA helix and impact on their structure. This was confirmed by a series of studies involving ctDNA molecules.10–17
In our previous papers, we have shown that phosphine derivatives of fluoroquinolone antibiotics (ciprofloxacin, norfloxacin, sparfloxacin) are characterized by a different biological activity than the parent drugs.18–20 The presence of the suitably located carboxyl and carbonyl groups in fluoroquinolones makes them ideal ligands, chelating various metal ions such as Mg2+, Ca2+, Cu2+, Zn2+, Co2+, Fe2+, Fe3+, Al3+.21–26 On the other hand the presence of a soft base phosphorus atom (in phosphine derivatives), offers a possibility of expanding the group of metals which can be coordinated i.e. Cu+, Pt2+, Pd2+, Au+, Ag+ and Ru2+.27–30
Exploring the dual nature of the phosphine-fluoroquinolone merger, we present herein copper(I) and (II) complexes with phosphine derivatives of ciprofloxacin or norfloxacin and diimine as an auxiliary ligand. Metal ion coordination to two different functional groups of the studied compounds should significantly impact the scope of action and could potentially generate interactions with cells through different mechanisms.
Results and discussion
In this work we present the physicochemical properties of four new complexes (see Fig. 1) and studies on their interactions with DNA. Two copper(I) complexes, ([CuI-PCp] and [CuI-PNr]), were obtained in the reaction of phosphine derivative of ciprofloxacin (PCp) or norfloxacin (PNr) with cupric iodide and 2,9-dimethyl-1,10-phenanthroline (dmp). The water soluble copper(II) complexes ([OPCp-CuII]+ and [OPNr-CuII]+) are the result of the reaction of oxide derivatives of PCp (OPCp) or PNr (OPNr) with [Cu(phen)(NO3)2]. | ||
| Fig. 1 Scheme of synthesis route of CuI and CuII complexes. | ||
The identities of all the compounds were confirmed using elemental analysis, 1H and 31P{H} NMR spectroscopy, mass spectrometry and X-ray techniques. An analysis of the MS spectra proved that both the aforementioned types of complexes have diverse stability. The copper(I) compounds are very labile, as evidenced by the lack of the molecular peak and the presence of the [Cu(dmp)]+ and [Cu(dmp)2]+ peaks. In the case of copper(II) complexes, molecular peaks can be observed, which indicates a greater stability of the chelates.
Spectroscopic methods
The analysis of the NMR spectra (CDCl3, r.t.) confirmed the identity and purity of the synthesized copper(I) complexes. Coordination of CuI by PCp or PNr causes changes in the chemical shift of the phosphorus atom in the 31P NMR spectrum analogous to those caused by the formation of chalcogenide derivatives.17–19 The signal of this atom for both complexes is widened and shifted towards lower fields by approx. 9 ppm (see Table S1 in ESI† for full details), similarly to the CuI complexes with other aminomethylphosphines.31,32 In the proton spectra, the most significant changes were observed for protons located close to the centre of coordination. A widened singlet of H1 proton from the Ph2PCH2– moiety is shifted towards lower fields and is placed at 3.59 ppm for [CuI-PNr], while free PNr gives this signal at 3.29 ppm (doublet, 2J(H1–P) = 2.75 Hz). For [CuI-PCp] this signal is considerably broadened and it was impossible to accurately determine its parameters. For both complexes, signals of H23 from the carboxylic group were noted. Their chemical shifts are equal to 15.06 ppm and 15.14 ppm, for [CuI-PCp] and [CuI-PNr], respectively. Signals of protons from dmp fragments prove the equivocal similarity of both complexes (Table S1, ESI†).Fluoroquinolones bear a few functional groups showing vibrations in the infrared region, therefore the IR spectra analysis of phosphines and their complexes is based on the most characteristic vibrations (vibration C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moiety of the carboxyl group and pyridone) in the 1730–1500 cm−1 range.12,33,34 Modifications of HCp or HNr leading to the formation of phosphines and their oxides do not change the positions of these bands, because they do not include carboxyl and pyridone groups. A similar situation can be observed upon formation of copper(I) complexes. On the contrary, the synthesis of copper(II) complexes significantly affects the vibrational energy of those groups. This is associated with the coordination of the metal by two oxygen atoms (one from the deprotonated carboxyl group and the other from the quinolone skeleton). A band at 1700–1730 cm−1, assigned to the CO stretching vibration of the carboxyl group, confirms that this group is protonated in the ligands of CuI complexes. For the parent drugs and their CuII complexes, this band is very weak or disappears,12,33 what confirms deprotonation of COOH. Additionally, in the case of copper(II) complexes the frequency of CO vibration of pyridone is shifted towards higher energies. These changes indicate that the CuII ion is coordinated via oxygen atoms from pyridone and carboxyl group. Nevertheless, we would like to emphasize that comparative analysis of the bands positions clearly demonstrates the structural conformity between phosphine derivatives and complexes of norfloxacin and ciprofloxacin in the solid state (see Fig. S1, ESI†).
O moiety of the carboxyl group and pyridone) in the 1730–1500 cm−1 range.12,33,34 Modifications of HCp or HNr leading to the formation of phosphines and their oxides do not change the positions of these bands, because they do not include carboxyl and pyridone groups. A similar situation can be observed upon formation of copper(I) complexes. On the contrary, the synthesis of copper(II) complexes significantly affects the vibrational energy of those groups. This is associated with the coordination of the metal by two oxygen atoms (one from the deprotonated carboxyl group and the other from the quinolone skeleton). A band at 1700–1730 cm−1, assigned to the CO stretching vibration of the carboxyl group, confirms that this group is protonated in the ligands of CuI complexes. For the parent drugs and their CuII complexes, this band is very weak or disappears,12,33 what confirms deprotonation of COOH. Additionally, in the case of copper(II) complexes the frequency of CO vibration of pyridone is shifted towards higher energies. These changes indicate that the CuII ion is coordinated via oxygen atoms from pyridone and carboxyl group. Nevertheless, we would like to emphasize that comparative analysis of the bands positions clearly demonstrates the structural conformity between phosphine derivatives and complexes of norfloxacin and ciprofloxacin in the solid state (see Fig. S1, ESI†).
Electronic spectra (Fig. S2, ESI†) of all the tested complexes were measured in DMSO (10% in H2O), due to their low solubility in water. Additionally, experiments repeated several times within 48 h demonstrated high stability of the studied compounds in solution. We observed characteristic bands with the maximum at ∼450 nm located in the typical range of the MLCT transition in the CuI complexes with dmp.31 In the spectra of the CuII complexes, in addition to the LMCT bands (∼190–350 nm) there are also d–d transitions observed. The differences in the coordination sphere around copper(II) ions between Cu(phen)(NO3)2 and [OPCp-CuII]+ as well as [OPNr-CuII]+ cause a hypsochromic shift of this band. In the case of [Cu(phen)(NO3)2], the band maximum is located around 709 nm, while for the complex with OPCp and OPNr – at around 649 and 631 nm, respectively (Fig. 2). Absorption increase resulting from the coordination of the copper(II) ion to phosphine derivative is probably associated with the molecular symmetry reduction.
 | ||
| Fig. 2 Electronic spectra of CuII compounds in range 575–900 nm. | ||
X-ray crystallography
Crystal structures were determined for both complexes of the HCp derivatives: [CuI-PCp] and [OPCp-CuII]+. X-ray structures of [CuI-PCp] in [CuI-PCp]·CH2Cl2·CH3CN and [OPCp-CuII]+ in [OPCp-CuII]NO3·3H2O are presented in Fig. 3 (solvent molecules and counter ion are omitted for clarity). | ||
| Fig. 3 X-ray structures of [CuI-PCp] and [OPCp-CuII]+. | ||
The coordination geometry of the cuprous ion in the [CuI-PCp] complex is a distorted tetrahedral geometry with a coordinating iodide ion, phosphine coordinating via P atom and dmp ligand chelating by two nitrogen atoms. Bond lengths (Cu1–I1: 2.559(1), Cu1–P1: 2.218(1), Cu1–N41: 2.130(2) and Cu1–N42: 2.068(2) Å) and angles (N41–Cu1–N42: 79.98(10), I1–Cu1–P1: 123.61(3)°) are in the range typical for CuI complexes with dmp and phosphines.35 However the angle between the plane of the dmp ligand and the plane of Cu1_I1_P1 atoms is only 83.10° indicating exceptionally large flattening deformations of the tetrahedral geometry. This may be caused by weak intramolecular π-stacking interactions between one of the phosphine –Ph rings (C61–C66) and the dmp ligand (the angle between the planes is 11.6° and the distance between the –Ph ring centroid and the dmp plane is 3.477 Å).
An asymmetric unit of the [OPCp-CuII]NO3·3H2O complex consists of the [OPCp-CuII]+ cation, a non-coordinating nitrate anion and three water molecules which do not form any strong hydrogen bonds. CuII ion is coordinated in equatorial plane to two strongly bound chelating ligands: phen molecule via nitrogen atoms and a deprotonated OPCp ligand via carboxylate and pyridone oxygen atoms. Bond lengths are as follows: Cu1–N41: 2.008(3), Cu1–N42: 2.014(3), Cu1–O17: 1.945(2) and Cu1–O23: 1.915(2) Å. The sum of the X–Cu–X angles equal to 358.6(1)° indicates an almost perfect square planar coordination mode.
Molecules of both complexes include two independent fused-ring fragments: the fluoroquinolone rings and the dmp or phen ligands. As it could be expected, this leads to interesting molecular packing of the complexes stabilized by π-stacking interactions. The molecular packing in the crystal of [CuI-PCp] presented in Fig. 4 is an example of a number of different intra- and intermolecular interactions involving π electrons of aromatic rings.36 Apart from the intramolecular π-stacking interactions between –Ph and dmp (Fig. 4A), several intermolecular interactions can be observed. The first of them are the interactions between the dmp rings and the rings of the –Cp moiety (Fig. 4B). They lead to the formation of centrosymmetric dimers linked by a double set of π-stacking interactions. The angle between the planes of the fused rings is 7.9° and the distances from the two Cp centroids to the dmp plane are 3.718 and 3.390 Å. Additionally, the dimers form a one-dimensional polymeric structure. It results from C–H⋯π interactions between the C61–C66 phenyl ring and C35 and H35 atoms of the C31–C36 phenyl ring from the neighboring dimer (Fig. 4C – dotted blue lines). The distances from the C61–C66 centroid to H35 and C35 are 2.718 and 3.602 Å, respectively. The angle C35–H35–π (C61–C66 centroid) is equal to 158.90°. The last type of the interactions is the π-stacking between –Cp fragments (Fig. 4D – the distance between the –Cp planes is equal to 3.594 Å).
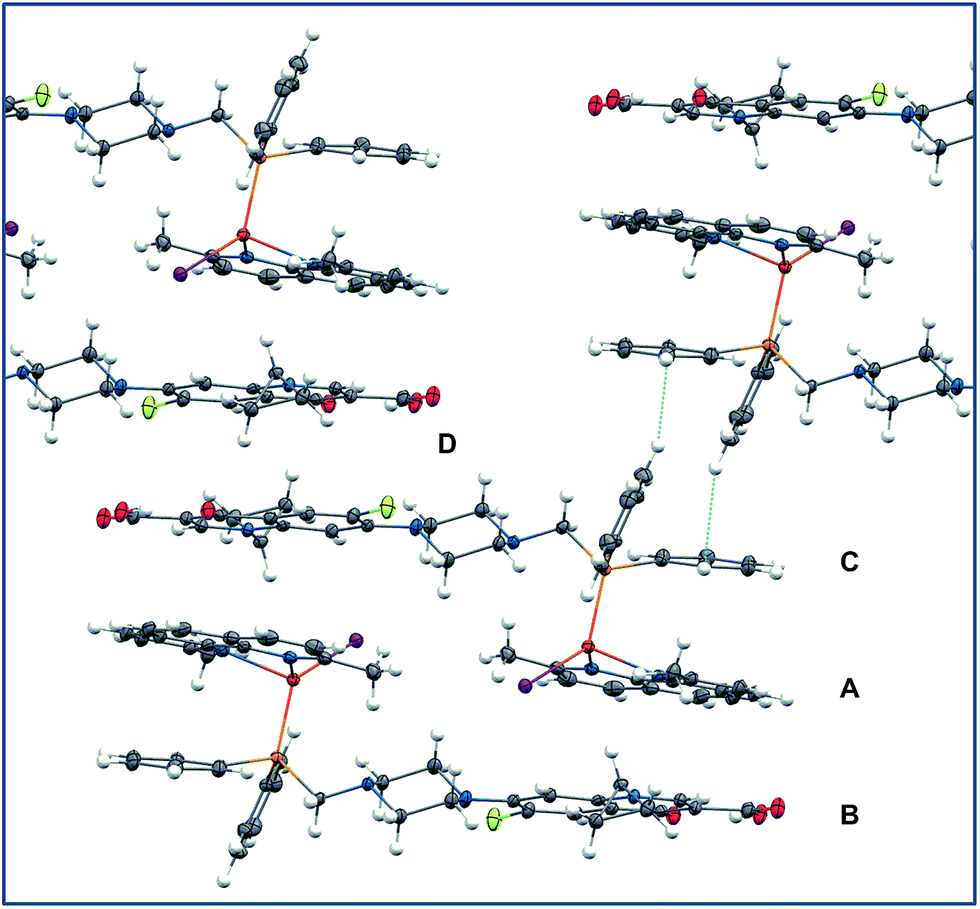 | ||
| Fig. 4 Molecular packing in the crystal of [CuI-PCp]. | ||
The analysis of the [OPCp-CuII]+ packing reveals that the coordination polyhedron around CuII ion is indeed tetragonally elongated octahedral of uniaxial symmetry (Fig. 5). CuII ion is additionally coordinated at axial positions by two oxygen atoms. The first Cu–O contact is found between Cu and pyridone oxygen from the neighboring complex molecule (Cu1–O17_b: 2.893(2) Å). As a result a dinuclear unit with two bridging O17 and O17_b atoms is formed with Cu1–Cu1_b distance of 3.752(1) Å, similarly to that observed for CuII complexes with other quinolones and aromatic diimines.37–40 The dinuclear unit is stabilized by a pair of π-stacking interactions between phen and –Cp fragments. The sixth coordination position around Cu1 is completed by a strong bond with O1_a atom from the OPPh2– fragment of the ligand involved in coordination to Cu1_a (r(Cu1–O1_a) = 2.243(2) Å). Hence two OPCp ligands attached simultaneously to Cu1 and Cu1_a act as the bridges in the second dinuclear system with the Cu–Cu1a distance of 14.622(1) Å. Both Cu1–O1_a and Cu1–O17_b bonds lead to the formation of one-dimensional coordination polymers.
 | ||
| Fig. 5 Molecular packing in the crystal of [OPCp-CuII]+. | ||
EPR
The powder EPR spectra for both studied CuII complexes are shown in Fig. 6a and c and compared with these for frozen water solutions (Fig. 6b and d). | ||
| Fig. 6 The EPR spectra of powder and water frozen solution of [OPNr-CuII]+ (a and b, respectively) and [OPCp-CuII]+ (c and d, respectively) together with the theoretical (simulated, sim b) spectrum giving the best fitting of the experimental spectra using the parameters presented in the text. The inserts show many times reinforced lines, experimental and simulated (sim b) due to ΔmS = 2 resonance transitions. The stars in the spectrum Fig. 6d indicate the signals due to mononuclear CuII species. | ||
In the case of [OPNr-CuII]+ the spectra Fig. 6a and b are similar in line shape, except that the hyperfine splitting is observed only for the frozen solution, Fig. 6b. The spectra correspond to the resonance transitions within S = 1 levels split in small degree in zero magnetic field, showing the ΔmS = 1 lines at low field (due to parallel orientation) and at low and high field (due to the perpendicular orientation), as well as in a weak forbidden line due to ΔmS = 2 (so-called half-field line). Additionally, the frozen solution spectrum exhibits copper hyperfine splitting consisting of seven lines, especially distinctly resolved for ΔmS = 2 line, what strongly proves that the electron spin is coupled with two copper nuclei, each having I = 3/2. The hyperfine interaction constant, Az = 86 G, corresponds to half the value for the monomeric CuN2O2 chromophore. All these observations confirm that both CuII ions are communicating, most likely in similar way as is revealed for solid state structure of [OPCp-CuII]+ (Fig. 5). The analogous interactions were observed for CuII complexes with other fluoroquinolones and N-donor heterocyclic ligands, but the Cu–Cu contacts were postulated as mediated only through π–π interactions between aromatic rings of N-donor heterocyclic ligands and quinolone molecules.37–40 In our opinion a weak exchange is also realized by CuII ions and pyridone oxygens (O17, Fig. 5) lying also in the parallel planes.
Although for [OPCp-CuII]+ complex, the powder EPR spectrum shows the ΔmS = 2 line, a dominant line is broad and isotropic (Fig. 6c). Taking into account that the frozen solution spectrum Fig. 6d exhibits the hyperfine splitting pattern due to two CuII nuclei, similar to that for [OPNr-CuII]+, the isotropic line in powder spectrum of [OPCp-CuII]+ consists of unresolved signals due to resonance transitions within S = 1 levels. The simultaneous simulation of S = 1 spectra, performed for both complexes, gave the best fit of theoretical spectrum (Fig. 6, sim b) to the experimental ones with g⊥ = 2.06, g‖ = 2.27; A⊥ = 20 G, A‖ = 85 G; |D| = 0.043 cm−1 and rotation of g tensor versus D tensor around Y axis by 18° for both complexes. Assuming only dipolar contributions to the zero-field splitting, the empirical formula based on the magnetic dipole-point model: Ddipole = −(g‖2 + 1/2g⊥2)μB2/r3, where D is in cm−1 and μB2 = 0.43297 in cm−1 Å−3, allowed to estimate the value of r (Cu–Cu distance) as about 4.2 Å, what is a value greater than that determined crystallographically (3.75 Å). The lower than expected dipolar contribution to the zero-field splitting is probably caused by delocalization of the unpaired electron into the ligand, not included in the dipole-point model.41
DNA strand break analysis
As DNA is the often target for antibiotics and anticancer drugs, studies on drug–DNA interactions play a key role in medicinal chemistry and pharmacology. Understanding of these processes is of prime significance in the rational design of more powerful, less toxic and more selective therapeutic. There are many different techniques for the studies of different aspects of the drug–DNA interactions.42For all synthesized compounds (organic ligands and complexes) we determined their interactions with deoxyribonucleic acid studying: strand break analysis using DNA plasmid, ability to intercalate into calf thymus DNA (ctDNA) and their binding modes with molecular docking.
The ability of complexes to induce single- and double-strand breaks was tested using gel electrophoresis of a DNA plasmid. A plasmid is a supercoiled (form I) molecule which can be easily separated from the open circular form (form II) as a result of single-stranded cleavage and from the linear form (form III) as a consequence of double-stranded cleavage of the sugar–phosphate backbone of the DNA chain. Due to the different shapes of all the three forms, they run in agarose gel at different velocities. This allows rapid assessment of the influence of the compounds on DNA.
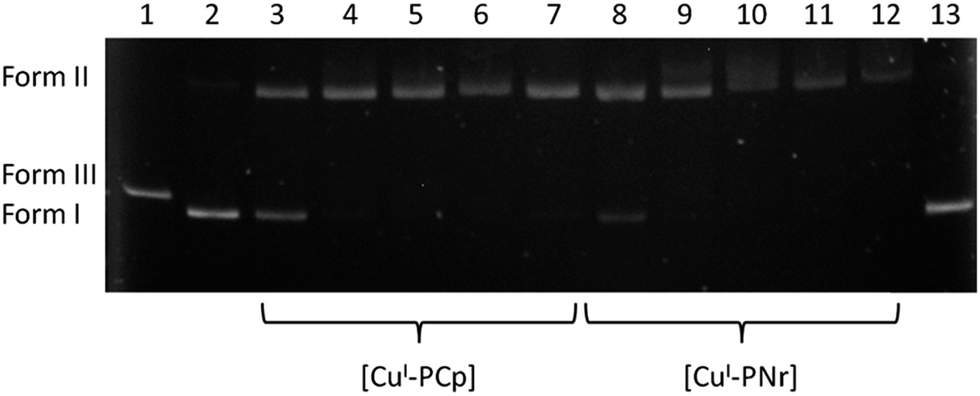
In the case of ligands (parent drugs and their phosphine derivatives) at the concentrations of up to 500 μM we did not observe any plasmid damage, only the native form was visible. However, the copper(I) complexes were able to generate damage in the plasmid (Fig. 7). The degree of DNA degradation was determined as a function of the complex concentration (10, 20, 50, 100 and 500 μM). An increase of the concentration results in the decrease of the native form amount and a simultaneous increase of the amount of form II. It should be noted that the latter form is observed even at the lowest concentrations of the complexes, which confirms the strong DNA-damaging ability of the tested compounds (lanes 3 and 8).
 | ||
| Fig. 7 Agarose gel electrophoresis of pBR322 plasmid cleavage by copper(I) complexes in a buffer solution (pH 7.4). Lanes: (1) plasmid linearized with EcoRI endonuclease; (2) plasmid – control; (3) +10 μM [CuI-PCp]; (4) +20 μM [CuI-PCp]; (5) +50 μM [CuI-PCp]; (6) +100 μM [CuI-PCp]; (7) +500 μM [CuI-PCp]; (8) +10 μM [CuI-PNr]; (9) +20 μM [CuI-PNr]; (10) +50 μM [CuI-PNr]; (11) +100 μM [CuI-PNr]; (12) +500 μM [CuI-PNr]; (13) plasmid + DMF. | ||
A densitometric analysis (Fig. 8) showed that [CuI-PNr] (lanes 8–12) degrades DNA more effectively in comparison with [CuI-PCp] (lanes 3–7). It is worth mentioning that a double-stranded DNA cleavage was not observed irrespective of the complex concentration. In the presence of H2O2, an endogenous oxidant, the studied complexes cause distinct changes in the plasmid structure (Fig. 9), resulting in a complete conversion of the supercoiled plasmid form to an open form at most concentrations.
 | ||
| Fig. 8 Densitometric analysis of plasmid cleavage by [CuI-PCp] (a) and [CuI-PNr] (b) (X axis: concentration [μM] on a logarithmic scale; Y axis: % plasmid forms). | ||
 | ||
| Fig. 9 Agarose gel electrophoresis of pBR322 plasmid cleavage by copper(I) complexes in a presence of 50 μM H2O2 in a buffer solution (pH 7.4). Lanes: (1) plasmid linearized with EcoRI endonuclease; (2) plasmid – control; (3) +10 μM [CuI-PCp] + H2O2; (4) +20 μM [CuI-PCp] + H2O2; (5) +50 μM [CuI-PCp] + H2O2; (6) +100 μM [CuI-PCp] + H2O2; (7) +500 μM [CuI-PCp] + H2O2; (8) +10 μM [CuI-PNr] + H2O2; (9) +20 μM [CuI-PNr] + H2O2; (10) +50 μM [CuI-PNr] + H2O2; (11) +100 μM [CuI-PNr] + H2O2; (12) +500 μM [CuI-PNr] + H2O2; (13) plasmid + DMF. | ||
Copper(II) complexes are not capable of double-helix damage at the tested concentrations, however the addition of H2O2 to the system leads to the formation of form II and form III. Simultaneously, the native form of the plasmid (I) completely disappears (Fig. 10 and 11). Similarly to the copper(I) complexes, [OPNr-CuII]+ causes a greater damage to the plasmid than [OPCp-CuII]+ (at lower concentrations, form II dominates and is more abundant).
 | ||
| Fig. 10 Agarose gel electrophoresis of pBR322 plasmid cleavage by copper(II) complexes in a presence of 50 μM H2O2 in a buffer solution (pH 7.4). Lanes: (1) plasmid – control; (2) +10 μM [OPCp-CuII]+ + H2O2; (3) +20 μM [OPCp-CuII]+ + H2O2; (4) +50 μM [OPCp-CuII]+ + H2O2; (5) +100 μM [OPCp-CuII]+ + H2O2; (6) +500 μM [OPCp-CuII]+ + H2O2; (7) +10 μM [OPNr-CuII]+ + H2O2; (8) +20 μM [OPNr-CuII]+ + H2O2; (9) +50 μM [OPNr-CuII]+ + H2O2; (10) +100 μM [OPNr-CuII]+ + H2O2; (11) +500 μM [OPNr-CuII]+ + H2O2. | ||
 | ||
| Fig. 11 Densitometric analysis of plasmid cleavage by [OPCp-CuII]+ (a) and [OPNr-CuII]+ (b) in a presence of 50 μM H2O2 (X axis: concentration [μM] on a logarithmic scale; Y axis: % plasmid forms). | ||
Interactions with calf thymus DNA (ctDNA)
The mechanism of drug interactions with the DNA helix may be based on the formation of covalent bonds. However, most substances interact with DNA noncovalently through three selective modes: (1) groove binding stabilized by hydrophobic, electrostatic and hydrogen-bonding interactions, (2) electrostatic binding to phosphate groups and (3) intercalation (inclusion of the heteroaromatic rings between complementary base pairs).43–45 The compounds presented in this paper include aromatic fused rings of the fluoroquinolone fragment and the diimine ligand in their structures, therefore they can act as potential intercalators. This assumption is supported by literature reports stating that unmodified fluoroquinolones are able to interact with DNA through electrostatic interactions with partial intercalation46,47 or are full intercalators.48We investigated the ability of ligands and complexes to intercalate DNA using ctDNA. Deoxyribonucleic acid from calf thymus is widely used in studies of DNA binding anticancer agents and other compounds that could modulate its structure and function. The ctDNA is a highly polymerized mixture of double (predominant) and single stranded DNA. The experiment was performed using fluorescence spectroscopy in the presence of ethidium bromide (EB), which is the most popular technique to study of the abovementioned processes. EB forms with ctDNA a water-soluble intercalation complex with strong fluorescence properties. The changes observed in the spectra of the ctDNA–EB complex, associated with the quenching of its luminescence, may indicate displacement of the intercalating EB and efficient interaction of the studied compound with the helix (Fig. S3 and S4, ESI†). The emission spectra of the ctDNA–EB system were recorded in the presence of increasing amounts of the compounds (0, 0.5; 1.0; 2.0; 5.0 and 10.0 molar ratio). Measurements were performed for 4 ciprofloxacin (PCp, OPCp, [CuI-PCp], [OPCp-CuII]+) and 4 norfloxacin (PNr OPNr, [CuI-PNr], [OPNr-CuII]+) derivatives and for unmodified drugs. Examination of the Stern–Volmer plots confirms that the fluorescence quenching of the ctDNA–EB complex by the tested compounds is in agreement with the linear equations (Fig. 12). The high structural similarity of both antibiotics, as well as their derivatives, suggests the same trends in the obtained results for both groups. However, an analysis of the obtained experimental data conclusively proved that HCp-based compounds displace EB much more efficiently than the analogous derivatives of HNr. Additionally, slightly different trends of the changes of activity among each group were observed. As regards ciprofloxacin and its derivatives, the activity follows the following order: PCp > [CuI-PCp] > [OPCp-CuII]+ ≫ HCp ≥ OPCp (Fig. 12a and S3, ESI†). As regards the norfloxacin derivatives, the intensity of the band decreases in the following order: [CuI-PNr] > [OPNr-CuII]+ > PNr ≫ OPNr ≥ HNr (Fig. 12b and S4, ESI†).
 | ||
| Fig. 12 Stern–Volmer plots of the ctDNA–EB system quenching by HCp derivatives (a) and HNr derivatives (b). (I0 and I – intensity of ctDNA–EB in the absence and presence of the increasing amounts of the studied compounds, respectively; n0 and n – mole numbers of the ctDNA–EB and the added compounds, respectively.) | ||
It should be noted that in case of a ten-fold excess of the phosphines, the emission intensity decreased by 51% for PCp and by 20% for PNr. The copper(I) complexes reduced the intensity of the band by 52% and 38% for [CuI-PCp] and [CuI-PNr] respectively, while in the case of the copper(II) complexes these changes were smaller: 30% for [OPCp-CuII]+ and 26% for [OPNr-CuII]+. Undoubtedly, fluorescence quenching of the ctDNA–EB system by the tested compounds indicates that these substances are able to displace EB from ctDNA and interact with the helix. Using the generally accepted theorem that any compound acts as an intercalator when its tenfold excess reduces the luminescence of the ctDNA–EB system by 50% we can, in principle, consider the studied compounds as partial intercalators.
Molecular docking
Molecular docking is a powerful method used mainly in the search of therapeutic agents designed to interact with a specific protein target. It allows to perform virtual screening on large libraries of compounds, rank the results, and propose structural hypotheses of how the ligands bind to the target.48–50 Docking is also of growing interest in the study of the binding of the chemical compounds to DNA.51–55 This method allows to investigate the interactions with short and well defined DNA sequences and give more insight into results obtained with experimental methods based on the studies of easily available but not well defined long DNA sequences. It is usually used to find the best orientation of ligand which would form a complex with a model of the DNA helix.Due to a large variety of the studied compounds, some of which carrying Cu+ or Cu2+ ions and a iodide anion, we employed the AutoDock Vina program, which is frequently used to study small molecule–DNA interactions. One of its advantages is an unique scoring function which does not use partial atomic charges.
Despite the usual exploitation of the DNA fragments from RCSB Protein Data Bank, we decided to create a B form of the DNA double-stranded hexadecanucleotide (sequence: ATATCGCGATATCGCG) with the geometry optimized using the AMBER ff99 force field.56 The length of this sequence exceeds a full helical turn and therefore it is possible to avoid investigations of ligand interactions with the deformed ends of the helix. Moreover, the chosen sequence is characterized by a clear distinction between fragments rich in AT and GC pairs. For the purpose of formation of the intercalation gaps we used a model molecule (tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine) which was inserted either between the 6th and 7th (G, C) base pairs (the “GC⋯GC” gap) or between the 10th and 11th (A, T) base pairs (the “AT⋯AT” gap) (Fig. S5, ESI†).
In order to test the DNA intercalating potential of the studied ligands and complexes, we investigated their interactions with the optimized hexadecanucleotide and both structures with pre-formed intercalating gaps. We also performed calculations for propidium iodide (PI) as well as for three model complexes: CuII complexes with ciprofloxacin or norfloxacin and phen ligand ([HCp-CuII]+ and [HNr-CuII]+) and CuI complex with dmp and P(CH2OH)Ph2 phosphine ([CuI-POH]). The geometries of the compounds were calculated using the DFT methods.
The geometry-optimized molecules were successfully docked onto the DNA segment. The qualitative analysis was based on the lowest energy poses for each molecule. The binding free energies [kcal mol−1] and remarks for all the tested compounds are given in Table 1. To show the different possible modes of interactions, in a few instances, data for two conformations are presented: (1) the lowest energy conformation (if not intercalative) and (2) the second lowest energy conformation (if intercalation was observed).
| B-DNA | B-(AT⋯AT) | B-(GC⋯GC) | ||||
|---|---|---|---|---|---|---|
| a a – [CuI-POH] ≡ [CuI(dmp){P(CH2OH)Ph2}]; b – placing in minor groove; c – major groove placing; Ø – no intercalation; p. i. – partial intercalation; f. i. – full intercalation. | ||||||
| HCp | −7.1 | (AT), b | −6.9 | p. i. | −7.7 | Ø |
| −7.4 | p. i. | |||||
| PCp | −8.4 | (GC), b | −8.0 | Ø | −7.3 | f. i. |
| OPCp | −8.6 | (AT), b | −7.9 | Ø | −8.1 | Ø |
| [CuI-PCp] | −7.9 | (AT), c | −7.8 | Ø | −7.8 | Ø |
| −7.7 | p. i. | |||||
| [OPCp-CuII]+ | −10.9 | (AT), b | −9.7 | Ø | −9.5 | f. i. |
| HNr | −7.1 | (GC), b | −7.7 | f. i. | −7.3 | p. i. |
| −7.2 | f. i. | |||||
| PNr | −8.4 | (GC), b | −7.9 | Ø | −8.0 | f. i. |
| OPNr | −8.5 | (AT), b | −7.9 | Ø | −8.3 | f. i. |
| −7.8 | f. i. | |||||
| [CuI-PNr] | −8.1 | (AT–GC), c | −9.0 | f. i. | −9.4 | f. i. |
| [OPNr-CuII]+ | −11.1 | (AT), b | −9.6 | Ø | −9.7 | f. i. |
| [HCp-CuII]+ | −8.7 | Ø | −10.0 | f. i. | ||
| −8.6 | p. i. | |||||
| [HNr-CuII]+ | −9.9 | f. i. | −9.9 | f. i. | ||
| [CuI–POH]a | −7.0 | Ø | −7.1 | p. i. | ||
| PI | −6.4 | −6.5 | f. i. | −6.5 | f. i. | |
The figures for all the interactions are presented in ESI (Fig. S6–S19, ESI†). Fig. 13 consists exemplary pictures for [CuI-PCp] and [CuI-PNr] complexes. Data analysis showed that a majority of compounds preferably bind to the minor grooves in the ATAT rather than in the GCGC region. Only CuI complexes ([CuI-PCp] and [CuI-PNr]) bind to major grooves. Interestingly, only in the case of HCp, HNr and [CuI-PNr] (as well as PI molecule) binding free energies were lower for intercalation modes of binding, than for the groove noncovalent binding. It showed that, in most cases, groove binding is energetically preferred over intercalation.
 | ||
| Fig. 13 Molecular docking: the docking of CuI complexes (with the scoring function results). | ||
Evaluation of the DNA intercalation revealed two general trends. First, the preferred binding site is the GC⋯GC fragment, as was demonstrated by the slightly lower binding energies. Second, the intercalation is more preferable for the HNr derivatives than for the HCp ones. It may be explained by the structural differences between cyclopropyl (HCp) and ethyl (HNr) substituent at N20 atom. As regards a majority of the HNr intercalating derivatives, –CH2CH3 chain remains in the ring plane which facilitates the insertion of the whole molecule between the base pairs. It is clearly contradictory to the experimental data obtained for the ctDNA–EB system, but at the same time it explains the higher efficiency of HNr complexes in the plasmid DNA cleavage.
Summing up, the molecular docking confirmed that the studied compounds interact with DNA by the groove binding as well as intercalation and showed the possible modes of interactions, what well supplements the obtained experimental results.
Experimental
Materials and methods
Reactions were carried out under a dinitrogen atmosphere using standard Schlenk techniques. CuI, 2,9-dimethyl-1,10-phenantroline (dmp), [Cu(phen)(NO3)2] and solvents were obtained from Sigma-Aldrich Co. All solvents were deaerated prior to use. PPh2CH2Cp (PCp), PPh2CH2Nr (PNr) and their oxide derivatives (OPCp and OPNr) were synthesized as described previously.17,18Syntheses
Yield 58%. Anal. calc. for C44H41CuIFN5O3P (928.2): C, 56.93; H, 4.45; N, 7.55. Found: C, 56.45; H, 4.52; N, 7.95%.
NMR (CDCl3, 298 K): 31P{1H}: −18.80 s; 1H: H23: 15.06 s, H19: 8.67 s, H4,7(dmp): 8.18 d J = 8.2, H13: 7.94 d J = 13.3, H5,6(dmp): 7.74 s, H3,8(dmp): 7.51 d J = 8.2, HPh(o),(m),(p): 7.42–7.17, H16: 7.42–7.17, H21: 3.56 m, H1: n.o, H2,6: 3.21, H15,16(dmp): 2.90 s, H3,5: 2.79, H22: 1.39 m, H22: 1.13 m.
MS (CDCl3): 479.1 [Cu(dmp)2]+ 100%; 271.0 [Cu(dmp)]+ 23%; 209.1 [(dmp)H]+ 4%; 663.1 0.2%.
Yield 63%. Anal. calc. for C43H41CuIFN5O3P (916.2): C, 56.36; H, 4.51; N, 7.65. Found: C, 55.93; H, 4.74; N, 7.76%.
NMR (CDCl3, 298 K): 31P{1H}: −18.14 s; 1H: H23: 15.14 s, H19: 8.62 s, H4,7(dmp): 8.19 d J = 8.2, H13: 8.00 d J = 13.2, H5,6(dmp): 7.76 s, H3,8(dmp): 7.52 d J = 8.2, HPh(o),(m),(p): 7.44–7.17, H16: 6.76 d J = 6.90, H21: 4.30 m, H1: 3.59 s, H2,6: 3.16, H15,16(dmp): 2.88 s, H3,5: 2.77, H22: 1.57 m.
MS (CDCl3): 480.1 [Cu(dmp)2H]+ 100%; 271.0 [Cu(dmp)]+ 3.7%; 669.0 [C30H32CuN5OP]+ 0.6%; 788.2 [Cu(dmp)PNr] 0.5%.
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3) solvent mixtures by slow evaporation.
:3) solvent mixtures by slow evaporation.Yield 78%. Anal. calc. for C42H36CuFN5O4P (788.3): C, 63.99; H, 4.57; N, 8.89. Found: C, 63.96; H, 4.59; N, 8.86%.
MS (MeOH): 568.2 [C27H25CuFN3O4]+ 100%; 293.1 [C13H9CuFNO2]+ 24.4%; 353.3 [C14H13CuFN3O3]+ 13.3%; 787.2 [M]+ 8.9%.
Yield 72%. Anal. calc. for C41H36CuFN5O4P (776.3): C, 63.43; H, 4.64; N, 9.02. Found: C, 63.40; H, 4.61; N, 9.01%.
MS (MeOH): 354.2 [C29H30FN3O4P]+ 100%; 332.1 [C17H19FN3O3]+ 10.7%; 775.2 [M]+ 3.6%.
General methods
Mass spectra were recorded on a Bruker Daltonics micrOTOF-Q mass spectrometer equipped with electrospray ionization (ESI) source and operated in positive ion mode. Elemental analysis was performed with a Vario EL3 CHN analyzer. IR spectra were recorded from 4000 to 400 cm−1 on Bruker 113v FTIR spectrophotometer as a KBr pellets.NMR spectra were recorded on a Bruker Avance 500 MHz spectrometer with traces of solvent as an internal reference for 1H spectra and 85% H3PO4 in H2O as an external standard for 31P. The spectra are defined as s = singlet (* – strongly broadened signal), d = doublet, and m = multiplet. Chemical shifts are reported in ppm and coupling constants are reported in Hz.
EPR spectra were recorded on a Bruker ELEXSYS E500 CW-EPR spectrometer equipped with an NMR teslameter (ER 036TM) and frequency counter at X-band frequency and at 77 K. The concentrations of the complexes were 1 × 10−3 M. The solutions were prepared using water and ethylene glycol (30 v/v%) as a cryoprotectant. The experimental spectra were simulated using spin (S = 1, I = 3/2) computer program written by Dr Andrew Ozarowski, National High Field Magnetic Laboratory, University of Florida.
Absorption spectra were recorded on a Cary 50 Bio spectrophotometer (Varian Inc., Palo Alto, CA) in the 1000–200 nm range, using 1 cm cuvettes. The concentration of the solutions was equal 5 × 10−5 M for [CuI-PCp] and [CuI-PNr]. For copper(II) complexes spectra were recorded in two concentrations: 5 × 10−5 M and 1 × 10−3 M.
X-ray crystallography
Single crystals of [CuI-PCp]·CH2Cl2·MeCN were obtained by slow crystallization of [CuI-PCp] from dichloromethane–acetonitrile solution at room temperature. Single crystals of [OPCp-CuII]NO3·3H2O were obtained analogously from water–acetonitrile solution.X-ray data were collected at 100 K using a KM4-CCD diffractometer and graphite-monochromated MoKα radiation generated from diffraction X-ray tube operated at 50 kV and 20 mA. The images were indexed, integrated, and scaled using the Oxford diffraction data reduction package.57 The structures were solved by direct methods (SHELXS Ver. 2013/1) and refined by the full-matrix least-squares method on all F2 data (SHELXL Ver. 2014/7).58 The data were corrected for absorption.57 Non H atoms were included in the refinement with anisotropic displacement parameters and the H atoms were included from geometry of the molecule or found in a difference Fourier map. Water hydrogen atoms in [OPCp-CuII]NO3·3H2O were found using CALC-OH utility59 within the WinGX v.2014.1 suite of programs60 and put into the structure unrefined with fixed positions.
Crystal/refinement data: [CuI-PCp]·CH2Cl2·MeCN ≡ C47H46Cl2CuFIN6O3P, Mr = 1054.21, crystal size: 0.12 × 0.10 × 0.07 mm, crystal system: triclinic, space group: P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , unit cell: a = 11.722(1) Å, b = 13.245(2) Å, c = 15.234(2) Å, α = 101.43(2)°, β = 99.33(1)°, γ = 94.55(1)°, V = 2272.4(5) Å3, Dcalcd(Z = 2) = 1.541 g cm−3, θ range for data collection: 2.88–36.95°, Mo Kα radiation (λ = 0.71073 Å), μMo = 1.365 mm−1, reflections collected/unique 43533/19449 [Rint = 0.0755], final R indices [I > 2σ(I)] R1 = 0.0562, wR2 = 0.0879, R indices (all data) R1 = 0.1515, wR2 = 0.1037, GOF = 0.867, largest diff. peak and hole: 0.903 and −1.243 e Å−3, data/restraints/parameters: 19449/0/563, T = 100(2) K.
, unit cell: a = 11.722(1) Å, b = 13.245(2) Å, c = 15.234(2) Å, α = 101.43(2)°, β = 99.33(1)°, γ = 94.55(1)°, V = 2272.4(5) Å3, Dcalcd(Z = 2) = 1.541 g cm−3, θ range for data collection: 2.88–36.95°, Mo Kα radiation (λ = 0.71073 Å), μMo = 1.365 mm−1, reflections collected/unique 43533/19449 [Rint = 0.0755], final R indices [I > 2σ(I)] R1 = 0.0562, wR2 = 0.0879, R indices (all data) R1 = 0.1515, wR2 = 0.1037, GOF = 0.867, largest diff. peak and hole: 0.903 and −1.243 e Å−3, data/restraints/parameters: 19449/0/563, T = 100(2) K.
Crystal/refinement data: [OPCp-CuII]NO3·3H2O ≡ C42H42CuFN6O10P, Mr = 904.32, crystal size: 0.12 × 0.07 × 0.06 mm, crystal system: triclinic, space group: P, unit cell: a = 12.174(2) Å, b = 13.613(1) Å, c = 14.483(1) Å, α = 62.36(1)°, β = 73.40(1)°, γ = 71.82(1)°, V = 1991.6(4) Å3, Dcalcd(Z = 2) = 1.508 g cm−3, θ range for data collection: 2.96–36.59°, Mo Kα radiation (λ = 0.71073 Å), μMo = 0.662 mm−1, reflections collected/unique 19259/9938 [Rint = 0. 0457], final R indices [I > 2σ(I)] R1 = 0.0579, wR2 = 0.1450, R indices (all data) R1 = 0.0938, wR2 = 0.1576, GOF = 0.969, largest diff. peak and hole: 0.835 and −0.826 e Å−3, data/restraints/parameters: 9938/0/550, T = 100(2) K.
DNA stand break analysis
The ability to induce strand breaks by the studied compounds was tested with the application of pBR322 plasmid. The solution of DNA in 50 mM sodium phosphate buffer (pH 7.4) was mixed with compounds. All compounds were dissolved in DMF, which concentration was kept constant (10% by volume) in the final solution. After 1 h incubations at 37 °C, reaction mixtures (20 μL) were mixed with 3 μL of loading buffer (bromophenol blue in 30% glycerol) and loaded on 1% agarose gels, containing ethidium bromide, in TBE buffer (90 mM Tris–borate, 20 mM EDTA, pH 8.0). Gel electrophoresis was done at constant voltage of 100 V (4 V cm−1) for 60 min. The gel was photographed and visualized with a Digital Imaging System (Syngen Biotech). For the densitometric analysis we used UltraQuant 6.0 program.Interactions with calf thymus DNA (ctDNA)
The stock solution was prepared by dissolving of ctDNA in the 50 mM phosphate buffer (pH = 7.4). The ctDNA concentration was determined by the analysis of a UV spectrum, using the molar absorption coefficient 6600 M−1 cm−1 at 258 nm. The stock solution was stored at 4 °C and used no more than 5 days. Luminescent complex of ctDNA with EB (ethidium bromide) was prepared by mixing of substrates in equimolar ratio (cctDNA–EB = 5 × 10−5 M; 50 mM of the phosphate buffer at pH = 7.4). The solution of the ctDNA–EB system was titrated by tested compounds (dissolved in DMSO) in different molar ratios (0.5, 1, 2, 5 and 10) and incubated for 1 h with any portion of the each compound. Photoluminescence measurements were recorded at 298 K on Cary Eclipse Fluorescence Spectrophotometer. Excitation wavelength was equal to 510 nm.Molecular docking
The structures of ligands (organic molecules and copper complexes) were optimized using the GAUSSIAN 09 package.61 We employed the DFT method using the hybrid functional of Truhlar and Zhao62 (M06). Basis set employed for geometry optimization calculations was: 6-31G* for all atoms (except I: 6-311G**63,64).The DNA models for docking were prepared according to the following procedure. Initial configuration of the canonical B form of the DNA double-stranded hexadecanucleotide (sequence: ATATCGCGATATCGCG) generated by the 3D-DART server,65 described by the AMBER ff99 force field,48 was optimized using the steepest descent method. No cutoffs for electrostatics or van der Waals terms were used due to a small system size; neither the periodic boundary conditions were used nor were the counterions added. Then, a previously optimized model intercalator molecule (tetrapyrido[3,2-a:2′,3′-c:3′′,2′′-h:2′′′,3′′′-j]phenazine) was inserted either between the 6th and 7th (C, G) base pairs (the “CG” gap) or between the 10th and 11th (A, T) base pairs (the “AT” gap). Such an insertion in the middle of the strand was devised to minimize the boundary effects. The intercalator was described by the GAFF force field, a generalized all-organic companion to the AMBER suite of force fields.66 The two intercalated structures were then subjected to energy minimization with the steepest descent procedure. Removal of the intercalator molecule produced two structures with intercalation gaps, used further in the docking studies. The molecular mechanics calculations were carried out with the GROMACS 4.5.5 software.67
For the docking analysis we used AutoDock Vina 1.1.2 program68 which applies a united-atom scoring function and ignores the user-supplied partial charges. Docking was performed as blind docking (it refers to the use of a box large enough to encompass any possible ligand–receptor interactions) with the set of boxes of 30 × 20 × 30 Å. Other parameters were set as follows: energy range = 10; exhaustiveness = 100. All structures for the docking experiments were prepared using AutoDock Tools 1.5.6 (ref. 69) and all single bonds in ligands were kept flexible during the experiments. The pictures were prepared using UCSF CHIMERA 1.9 (ref. 70) and POV-Ray 3.7.0.71
Conclusions
Four copper complexes with phosphine derivatives of the 2nd generation quinolone antibiotics, ciprofloxacin or norfloxacin, were synthesized. The identities and structures of the obtained new compounds were confirmed by a series of spectroscopic techniques. X-ray spectroscopy allowed to determine the solid state structures of the two complexes with the HCp derivatives: ([CuI-PCp] and [OPCp-CuII]+). The metal ion in the CuI complexes is coordinated by P atom from PCp or PNr, an iodide ion and two N atoms from dmp ligand. On the other hand, the metal ion in the CuII complexes is coordinated by the quinolone fragment of OPCp or OPNr (chelating via carboxylate and pyridone oxygen atoms) and a phen molecule (via two nitrogen atoms). [OPCp-CuII]+ in the solid state forms dimers with Cu atoms bound by pyridone oxygen bridges. Interestingly, the dimeric structures also form in the solutions, what was confirmed by the analysis of the EPR spectra.We determined the interactions of all the synthesized compounds with deoxyribonucleic acid studying: plasmid DNA strand break analysis, their ability to insert between the DNA bases and the possible binding modes.
The analysis of the gel electrophoresis data showed that, unlike the uncoordinated ligands, the complexes caused DNA degradation. In general, the Cu(I) and Cu(II) complexes with HCp based derivatives cleaved the plasmid less efficiently than the analogous compounds with the HNr moiety. Copper(I) complexes were able to induce only single-strand breaks of the sugar–phosphate backbone of the DNA chain. An increase of their concentration resulted in an increase of the open-circular form (II) amount. The addition of H2O2 to the system resulted in a more efficient formation of form II, as a consequence of single strand breaking, and the complete disappearance of form I. Copper(II) complexes were also able to damage the plasmid but only in the presence of an oxidant. For these complexes, the native form of the plasmid completely disappeared, while the II and III (linear) forms were observed.
The investigations of the intercalation of the studied compounds with ctDNA showed that the HCp derivatives and their complexes were characterized by a higher ability to interact with the helix than HNr derivatives. Moreover, unmodified antibiotics and the oxide derivatives interacted more weakly with ctDNA in comparison with the phosphines and the complexes, which were able to partially intercalate between the complementary base pairs.
The performed molecular docking calculations showed that the most of the studied compounds preferably bind to the minor or major ([CuI-PCp] and [CuI-PNr] only) grooves of the DNA double helix. They are also able to intercalate the into GC⋯GC fragment as the preferred site. Interestingly, the intercalation was more preferable for the HNr derivatives than for the HCp ones, which was contradictory with the data obtained for the ctDNA, but simultaneously confirmed the higher efficiency of the HNr complexes in the plasmid cleavage.
It is recognized that copper complexes are not only promising therapeutics with an anticancer activity, but also potential antimicrobial agents. The biological activity of the complexes presented in this paper could increase or remain unchanged compared to parent drugs, but in both cases large structural modifications may overcome the antibiotic resistance gained by microorganisms. Now, we are focusing on in vitro studies of antimicrobial activity of the compounds presented herein towards several bacterial strains and investigations of their cytotoxicity towards selected tumor cell lines.
Acknowledgements
Authors would like to thank Dr Jarosław Panek for the valuable help with the generation of the DNA hexadecanucleotide models. Financial support from the Polish National Science Centre (grant 2011/03/B/ST5/01557) is gratefully acknowledged. DFT calculations have been carried out in Wroclaw Centre for Networking and Supercomputing (http://www.wcss.wroc.pl) – grant No. 140.Notes and references
- P. C. A. Bruijnincx and P. J. Sadler, Curr. Opin. Chem. Biol., 2008, 12, 197–206 CrossRef CAS PubMed.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089–1097 CrossRef CAS PubMed.
- C. X. Zhang and S. J. Lippard, Curr. Opin. Chem. Biol., 2003, 7, 481–489 CrossRef CAS PubMed.
- K. H. Thomson and C. Orvig, Dalton Trans., 2000, 2885–2892 RSC.
- M. J. Pucci and K. Bush, Clin. Microbiol. Rev., 2013, 26, 792–821 CrossRef CAS PubMed.
- V. Uivarosi, Molecules, 2013, 18, 11153–11197 CrossRef CAS PubMed.
- L. J. Ming, Med. Res. Rev., 2003, 23, 697–762 CrossRef CAS PubMed.
- J. Nagaj, P. Kołkowska, A. Bykowska, U. K. Komarnicka, A. Kyzioł and M. Jeżowska-Bojczuk, Med. Chem. Res., 2015, 24, 115–123 CrossRef CAS PubMed.
- K. Stokowa-Sołtys, N. Gaggelli, J. Nagaj, W. Szczepanik, J. Ciesiołka, J. Wrzesiński, A. Górska, E. Gaggelli, G. Valensin and M. Jeżowska-Bojczuk, J. Inorg. Biochem., 2013, 124, 26–34 CrossRef PubMed.
- R. Singh, A. Debnath, D. T. Masram and D. Rathore, Res. J. Chem. Sci., 2013, 3, 83–94 CAS.
- C. A. Akinremi, J. A. Obaleye, S. A. Amolegbe, J. F. Adediji and M. O. Bamigboye, Int. J. Med. Biomed. Res., 2012, 1, 24–34 CrossRef.
- W. Szczepanik, J. Ciesiołka, J. Wrzesiński, J. Skała and M. Jeżowska-Bojczuk, Dalton Trans., 2003, 1488–1494 RSC.
- W. Szczepanik, E. Dworniczek, J. Ciesiołka, J. Wrzesiński, J. Skała and M. Jeżowska-Bojczuk, J. Inorg. Biochem., 2003, 94, 355–364 CrossRef CAS PubMed.
- K. Stokowa-Sołtys, A. Kasprowicz, J. Wrzesiński, J. Ciesiołka, N. Gaggelli, E. Gaggelli, G. Valensin and M. Jeżowska-Bojczuk, J. Inorg. Biochem., 2015 DOI:10.1016/j.jinorgbio.2015.05.011.
- T. Bortolotto, P. Pereira Silva, A. Neves, E. C. Pereira-Maia and H. Terenzi, Inorg. Chem., 2011, 50, 10519–10521 CrossRef CAS PubMed.
- J.-B. Chao, M.-D. Xu, C.-X. Yin and S.-P. Huang, Biochemistry, 2007, 72, 153–161 CAS.
- B. S. Sekhon, J. Pharm. Educ. Res., 2010, 1, 1–20 Search PubMed.
- A. Bykowska, R. Starosta, A. Brzuszkiewicz, B. Bażanow, M. Florek, N. Jackulak, J. Król, J. Grzesiak, K. Kaliński and M. Jeżowska-Bojczuk, Polyhedron, 2013, 60, 23–29 CrossRef CAS.
- A. Bykowska, R. Starosta, U. K. Komarnicka, Z. Ciunik, A. Kyzioł, K. Guz-Regner, G. Bugla-Płoskońska and M. Jeżowska-Bojczuk, New J. Chem., 2014, 38, 1062–1071 RSC.
- U. K. Komarnicka, R. Starosta, K. Guz-Regner, G. Bugla-Płoskońska, A. Kyzioł and M. Jeżowska-Bojczuk, J. Mol. Struct., 2015, 1096, 55–63 Search PubMed.
- P. Živec, F. Perdih, I. Turel, G. Giester and G. Psomas, J. Inorg. Biochem., 2012, 117, 35–47 CrossRef PubMed.
- G. Psomas, J. Inorg. Biochem., 2008, 102, 1798–1811 CrossRef CAS PubMed.
- M. J. Feio, I. Sousa, M. Ferreira, L. Cunha-Silva, R. G. Saraiva, C. Queirós, J. G. Alexandre, V. Claro, A. Mendes, R. Ortiz, S. Lopes, A. L. Amaral, J. Lino, P. Fernandes, A. J. Silva, L. Moutinho, B. de Castro, E. Pereira, L. Perelló and P. Gameiro, J. Inorg. Biochem., 2014, 138, 129–143 CrossRef CAS PubMed.
- M. P. López-Gresa, R. Ortiz, L. Perelló, J. Latorre, M. Liu-González, S. García-Granda, M. Pérez-Priede and E. Cantón, J. Inorg. Biochem., 2002, 92, 65–74 CrossRef.
- M. N. Patel, M. R. Chhasatia, P. A. Dosi, H. S. Bariya and V. R. Thakkar, Polyhedron, 2010, 29, 1918–1924 CrossRef CAS.
- Y. Wang, G.-W. Lin, J. Hong, L. Li, Y.-M. Yang and T. Lu, J. Coord. Chem., 2010, 63, 3662–3675 CrossRef CAS.
- C. Marzano, V. Gandin, M. Pellei, D. Colavito, G. Papini, G. G. Lobbia, E. Del Giudice, M. Porchia, F. Tisato and C. Santini, J. Med. Chem., 2008, 51, 798–808 CrossRef CAS PubMed.
- J. J. Liu, P. Galettis, A. Farr, L. Maharaj, H. Samarasinha, A. C. McGechan, B. C. Baguley, R. J. Bowen, S. J. Berners-Price and M. J. McKeage, J. Inorg. Biochem., 2008, 102, 303–310 CrossRef CAS PubMed.
- F. R. Pavan, G. V. Poelhsitz, L. V. P. da Cunha, M. I. F. Barbosa, S. R. A. Leite, A. A. Batista, S. H. Cho, S. G. Franzblau, M. S. de Camargo, F. A. Resende, E. A. Varanda and C. Q. F. Leite, PLoS One, 2013, 8, 1–10 Search PubMed.
- R. Starosta, A. Bykowska, M. Barys, A. K. Wieliczko, Z. Staroniewicz and M. Jeżowska-Bojczuk, Polyhedron, 2011, 30, 2914–2921 CrossRef CAS.
- R. Starosta, U. K. Komarnicka and M. Puchalska, J. Lumin., 2014, 145, 430–437 CrossRef CAS.
- R. Starosta, M. Puchalska, J. Cybińska, M. Barys and A. V. Mudring, Dalton Trans., 2011, 40, 2459–2468 RSC.
- V. L. Dorofeev, Pharm. Chem. J., 2004, 38, 693–697 CrossRef CAS.
- A. K. Chattah, Y. Garro Linck, G. A. Monti, P. R. Levstein, S. A. Breda, R. H. Manzo and M. E. Olivera, Magn. Reson. Chem., 2007, 45, 850–859 CrossRef CAS PubMed.
- R. Starosta, M. Puchalska, J. Cybińska, M. Barys and A. V. Mudring, Dalton Trans., 2011, 40, 2459–2468 RSC.
- C. Janiak, J. Chem. Soc., Dalton Trans., 2000, 3885–3896 RSC.
- E. K. Efthimiadou, N. Katsaros, A. Karaliota and G. Psomas, Inorg. Chim. Acta, 2007, 360, 4093–4102 CrossRef CAS.
- E. K. Efthimiadou, H. Thomadaki, Y. Sanakis, C. P. Raptopoulou, N. Katsaros, A. Scorilas, A. Karaliota and G. Psomas, J. Inorg. Biochem., 2007, 101, 64–73 CrossRef CAS PubMed.
- E. K. Efthimiadou, Y. Sanakis, M. Katsarou, C. P. Raptopoulou, A. Karaliota, N. Katsaros and G. Psomas, J. Inorg. Biochem., 2006, 100, 1378–1388 CrossRef CAS PubMed.
- G. Psomas, A. Tarushi, E. K. Efthimiadou, Y. Sanakis, C. P. Raptopoulou and N. Katsaros, J. Inorg. Biochem., 2006, 100, 1764–1773 CrossRef CAS PubMed.
- D. L. Reger, A. Debreczeni, M. D. Smith, J. Jezierska and A. Ożarowski, Inorg. Chem., 2012, 51, 1068–1083 CrossRef CAS PubMed.
- A. Mukherjee and W. D. Sasikala, Adv. Protein Chem. Struct. Biol., 2013, 92, 1–62 CAS.
- P. Zivec, F. Perdih, I. Turel, G. Giester and G. Psomas, J. Inorg. Biochem., 2012, 117, 35–47 CrossRef CAS PubMed.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 28, 4565–4579 RSC.
- P. Drevensek, N. Poklar Ulrih, A. Majerle and I. Turel, J. Inorg. Biochem., 2006, 100, 1705–1713 CrossRef CAS PubMed.
- I. Abu-Shqair, N. Diab, R. Salim and M. Al-Subu, Asian J. Appl. Sci., 2013, 1, 179–184 Search PubMed.
- X. Linga, W. Zhonga and H. Kunyi, J. Photochem. Photobiol., B, 2008, 93, 172–176 CrossRef PubMed.
- A. Tarushi, E. Polatoglou, J. Kljun, I. Turel, G. Psomas and D. P. Kessissoglou, Dalton Trans., 2011, 40, 9461–9473 RSC.
- Drug Discovery Research: New Frontiers in the Post-Genomic Era, ed. Z. Huang, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2007 Search PubMed.
- A. Hernández-Santoyo, A. Y. Tenorio-Barajas, V. Altuzar, H. Vivanco-Cid and C. Mendoza-Barrera, in Technology and Application, ed. T. Ogawa, InTech, Rijeka, Croatia, 2013 Search PubMed.
- H. Ye, C. Cande, N. C. Stephanou, S. Jiang, S. Gurbuxani, N. Larochette, E. Daugas, C. Garrido, G. Kroemer and H. Wu, Nat. Struct. Biol., 2002, 9, 680–684 CrossRef CAS PubMed.
- A. Srishailam, Y. P. Kumar, P. V. Reddy, N. Nambigari, U. Vuruputuri, S. S. Singh and S. Satyanarayana, J. Photochem. Photobiol., B, 2014, 132, 111–123 CrossRef CAS PubMed.
- R. Gaur, R. A. Khan, S. Tabassum, P. Shah, M. I. Siddiqi and L. Mishra, J. Photochem. Photobiol., A, 2011, 220, 145–152 CrossRef CAS.
- D. Shao, M. Shi, Q. Zhao, J. Wen, Z. Geng and Z. Wang, Z. Anorg. Allg. Chem., 2015, 641, 454–459 CrossRef CAS.
- T. R. Arun, R. Subramanian, S. Packianathan and N. Raman, J. Fluoresc., 2015, 25, 1127–1140 CrossRef CAS PubMed.
- M. Wang, P. Cieplak and P. A. Kollman, J. Comput. Chem., 2000, 21, 1049–1074 CrossRef.
- Oxford Diffraction, CRYSALIS–CCD and CRYSALIS–RED, Oxford Diffraction Ltd, Abingdon, England, 2010 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 47, 112–122 CrossRef PubMed.
- M. Nardelli, J. Appl. Crystallogr., 1999, 32, 563–571 CrossRef CAS.
- L. J. Farrugia, J. Appl. Crystallogr., 2012, 45, 849–854 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 Search PubMed.
- D. Feller, J. Comput. Chem., 1996, 17, 1571–1586 CrossRef CAS.
- K. L. Schuchardt, B. T. Didier, T. Elsethagen, L. Sun, V. Gurumoorthi, J. Chase, J. Li and T. L. Windus, J. Chem. Inf. Model., 2007, 47, 1045–1052 CrossRef CAS PubMed.
- M. van Dijk and A. M. J. J. Bonvin, Nucleic Acids Res., 2009, 37, 235–239 CrossRef PubMed.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- S. Pronk, S. Páll, R. Schulz, P. Larsson, P. Bjelkmar, R. Apostolov, M. R. Shirts, J. C. Smith, P. M. Kasson, D. van der Spoel, B. Hess and E. Lindahl, Bioinformatics, 2013, 29, 845–854 CrossRef CAS PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 CAS.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 16, 2785–2791 CrossRef PubMed.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
- POV-Ray for Windows, v.3.7.0, http://www.povray.org.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1053098 and 1053099. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra07483e |
| This journal is © The Royal Society of Chemistry 2015 |