
Self-assembled core–shell nanospheres and dendritic nanostructure of novel tetra-(3-phenyprop-2-allyloxy) phthalocyanine in different solvents†
Abstract
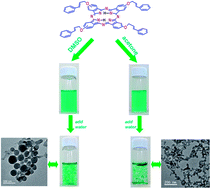
Novel metal-free tetra-(3-phenyprop-2-allyloxy) phthalocyanine (TPAO-Pc) was synthesized and characterized by nuclear magnetic resonance spectrum (1H NMR), MALDI-TOF MS, Fourier-transform infrared (FT-IR) and UV-Visible absorption (UV-Vis) spectra. The effects of concentrations and solvents on the properties of self-assembly were investigated via absorption and fluorescence spectra, transmission electron microscopy (TEM). TPAO-Pc was stable in most organic solvents, such as toluene, acetone and dimethylsulfoxide (DMSO). However, the TPAO-Pc exhibited the tendency of forming “face-to-face” stacking mode self-assemblies (H-aggregate) in DMSO–water and acetone–water mixed solutions. Self-assembly of the TPAO-Pc was also demonstrated by fluorescence spectra. The TEM images displayed the core–shell nanospheres and dendritic nanostructure of TPAO-Pc formed in the DMSO–water and acetone–water mixed solutions, respectively. The mechanism of the self-assemblies growth was proposed on the basis of the experimental results. The morphology of the TPAO-Pc self-assemblies was related with the selected solvent conditions and the aggregation time. H-bond, π–π interaction may be the main driving force for the formation of core–shell nanospheres and dendritic nanostructure.
 Please wait while we load your content...
Please wait while we load your content...

