
Charge heterogeneity induced binding and phase stability in β-lacto-globulin–gelatin B gels and coacervates at their common pI†
Abstract
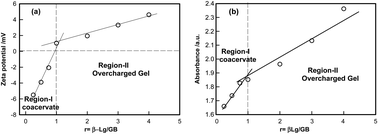
An understanding of the interactions between gelatin B (GB) and β-lacto-globulin (β-Lg) mainly arising from surface selective patch binding occurring at their common pI (≈5.0 ± 0.5) in the absence of added salt. Heterogeneous surface charge distribution on β-Lg facilitated such interaction at different mixing ratio ([GB]: [β-Lg] = r) and the GB–β-Lg complexes carried distinctive surface charge (seen through their zeta potential, ζ). For r < 1 : 1 (partial charge neutralization, ζ ≈ 0) a turbid solution was formed which gives the indication of formation of intermolecular soluble complexes. For r > 1 : 1 (overcharged regime, ζ > 0) the dispersion remained transparent and homogeneous which gives no phase separation, but the dispersion formed a gel on waiting. The overcharged gels were homogeneous, more rigid and higher melting temperature in comparison to coacervate. In the coacervate phase, the intensity of the scattered light Is, and its time-correlation function [g2(t) − 1] did not evolve with time. In contrast, the gel phase displayed considerable change with aging time tw. For gels, as tw → ∞ the system moved from an ergodic to non-ergodic state. At tw = 0, the correlation function exhibited one relaxation mode due to the system residing deeply inside the ergodic phase and purely mirroring Brownian dynamics. After a characteristic waiting time, tw an additional mode (slow relaxation) appeared which was attributed to inter-chain interaction induced reorganization of entanglements. This characteristic time was the time required for the system to get dynamically arrested, similar observation was made from rheology measurements too. A comprehensive phase diagram depicting the stability of the dispersion in various charged soft matter states of the complex under various temperature conditions was established.
 Please wait while we load your content...
Please wait while we load your content...

