
Efficient bifunctional materials based on pyrene- and triphenylamine-functionalized dendrimers for electroluminescent devices†
T. Keawina,
N. Prachumrakb,
S. Namuangrukc,
S. Pansaya,
N. Kungwand,
S. Maensirie,
S. Jungsuttiwonga,
T. Sudyoadsukb and
V. Promarak*b
aDepartment of Chemistry, Faculty of Science, Ubon Ratchathani University, Ubon Ratchathani 34190, Thailand
bSchool of Molecular Science and Engineering, Vidyasirimedhi Institute of Science and Technology, Wangchan, Rayong, 21210, Thailand. E-mail: vinich.p@vistec.ac.th
cNational Nanotechnology Center, 130 Thailand Science Park, Klong Luang, Pathumthani 12120, Thailand
dChemistry Department, Faculty of Science, Chiang Mai University, Chiang Mai 50200, Thailand
eSchool of Physics, Institute of Science, Suranaree University of Technology, Muang District, Nakhon Ratchasima 30000, Thailand
First published on 24th August 2015
Abstract
To realize highly efficient bifunctional blue-light emitting and hole-transporting materials for OLEDs, a series of pyrene- and triphenylamine-peripheral functionalized carbazole dendrimers, namely G1PYR, G2PYR, G1TPA and G2TPA, were designed, synthesized and characterized. Especially, G2PYR having four pyrene units substituted on the 2nd generation carbazole dendritic scaffold exhibited a strong blue emission with high Tg amorphous and good film-forming properties. Simple structured blue OLED (λEL = 463 nm) using G2PYR as emissive layer and Alq3-based green OLED (λEL = 512 nm) using G2PYR as hole-transporting layer with high luminance efficiencies (η) and low turn-on voltages (Von) of 5.89 cd A−1 and 3.1 V, and 5.15 cd A−1 and 2.6 V were attained, respectively.
Introduction
In the last decade, we have seen great efforts taken in the field of organic light-emitting diodes (OLEDs) to develop new materials as well as to optimize device fabrication conditions in order to realize commercial applications in full colour displays and solid-state lightings with high efficiency and long lifetime.1 Among the established light-emitting (EL) materials, only red and green systems have shown satisfactory efficiencies, colour purity and lifetimes to be of commercial value. Because of the large band gap energy, the performance of blue electroluminescent emitters is usually inferior to those of green and red emitters. Despite many blue electroluminescent systems, including fluorenes and spirofluorenes,2 metal complexes,3 oxadiazoles,4 distyrylarylene derivatives,5 pyrenes,6 anthracenes,7 aromatic amines,8 and heterocyclic compounds,9 have been synthesized and investigated, developing blue-emitters exhibiting not only with high efficiency but also with simple fabrication method remains a key challenge. Particularly, pyrene-based blue fluorescent emitters have attracted large attention for OLEDs, because the photoluminescence quantum yield, carrier mobility, and the electron-injection ability of emitters made of pyrene are higher when compared to those of fluorene derivatives.10 However, a weakness of pyrene is that its emission in the solid state is effectively suppressed due to the formation of excimers via π–π stacking.11 Numerous efforts have been attempted to improve the photophysical properties of pyrene such as 3,6-dipyrenylcarbazole end capped oligofluorenes,12 tetraarylpyrenes13 and 9,9-bis-(3-(9-phenyl-carbazoyl))-2,7-dipyrenylfluorene,14 pyrene-1,3-alt-calix[4]arene,15 fluorene-substituted pyrenes,16 2,7-bispyrene-9,9-bis(4-diphenylaminophenyl)fluorenes,17 pyrene-modified carbazole oligomers18 and pyrene functionalized octavinylsilsesquioxane cores.19 In terms of the device fabrication, solution-processed OLEDs fabricated using molecular amorphous materials will have great advantages, because the materials used are easy to synthesize and purify, while the fabrication method is convenient, low cost and allows large-scale manufacturing with less material usage.20 Several chemical approaches have been investigated for the development of such thermally stable amorphous EL materials.21 These lead to our design of new molecular materials combining the fine photoluminescent efficiency and electron-transporting ability of pyrene units22 with the hole-transporting capability, high thermal stability and glass state-forming ability of carbazole dendritic scaffolds.23 We deduced that the sterically congested carbazole dendritic platforms24 in these molecules would avert the peripheral pyrene rings to undesirable self-quenching in the solid state as well as improve the physical properties of the molecule, thereby realizing pyrene systems with pure blue-light emitting and hole-transporting bifunctional aspects for simple structured solution-processed OLEDs.Herein we report a detailed synthesis of a series of pyrene- and triphenylamine-functionalized carbazole dendrimers (Scheme 1) as well as their physical and photophysical properties. The investigation of the device fabrication and performance using these materials as an emissive layer and hole-transporting layer is also reported.
 | ||
| Scheme 1 Synthesis of GnPYR and GnTPA. | ||
Results and discussion
Materials synthesis
Scheme 1 outlines the synthesis of the bifunctional pyrene- and triphenylamine-peripheral functionalized carbazole dendrimers, namely GnPYR (G1PYR and G2PYR) and GnTPA (G1TPA and G2TPA). Ullmann coupling of an available 3,6-dibromocarbazole 1 with carbazole (2.2 equiv.) in the presence of CuI/±trans-1,2-diaminocyclohexane as a catalyst K3PO4 as a base in toluene followed by bromination of the resultant 2 with NBS in THF afforded the tetrabromo 2nd generation carbazole dendritic platform 3 in a good yield. Suzuki cross coupling of the bromide scaffolds 1 and 3 with the excess amount of either pyrene-1-boronic acid or 4-(diphenylamino)phenylboronic acid catalyzed by Pd(PPh3)4/2 M Na2CO3 (aq.) in THF gave the target G1PYR (light green solid), G2PYR (light green solid), G1TPA (white solid) and G2TPA (white solid) in 85–91% yields. The structures and purities of the compounds were confirmed by 1H NMR, 13C NMR and MALDI-TOF MS. These newly synthesized dendrimers show good solubility in most organic solvents ensuring that their thin films could be fabricated by low-cost solution casting processes.The quality and morphology of the spin-casting film, which is one of the key important factors for the OLEDs fabrication and performance, was exanimated by atomic force microscopy (AFM). Fig. 1 displays the tapping mode AFM images of the films spin-coated from CHCl3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :toluene solution of GnPYR and GnTPA. The AFM images of thin films of the 2nd generation dendrimers (G2PYR and G2TPA) show a very uniform and smooth surface, indicating excellent film-forming properties, while the surface of the spin-coated films of the 1st generation dendrimers (G1PYR and G1TPA) is a little coarser. These AFM images give evidence that the film uniformity is mostly determined by the generation or size of the dendrimers and in high-generation the dendrimer branches can relatively easily interpenetrate each other and establish intermolecular entanglements. Particularly, the spin-coated films of G2PYR and G2TPA are subjected to heating at 100 °C and inspected by AFM periodically. The films remain unchanged after several hours. This is very important for the emissive materials to be thermally stable amorphous to avoid grain-boundary defects, which could reduce the efficiency of the OLEDs by hindering a charge migration.25
:toluene solution of GnPYR and GnTPA. The AFM images of thin films of the 2nd generation dendrimers (G2PYR and G2TPA) show a very uniform and smooth surface, indicating excellent film-forming properties, while the surface of the spin-coated films of the 1st generation dendrimers (G1PYR and G1TPA) is a little coarser. These AFM images give evidence that the film uniformity is mostly determined by the generation or size of the dendrimers and in high-generation the dendrimer branches can relatively easily interpenetrate each other and establish intermolecular entanglements. Particularly, the spin-coated films of G2PYR and G2TPA are subjected to heating at 100 °C and inspected by AFM periodically. The films remain unchanged after several hours. This is very important for the emissive materials to be thermally stable amorphous to avoid grain-boundary defects, which could reduce the efficiency of the OLEDs by hindering a charge migration.25
 | ||
| Fig. 1 Tapping mode AFM images of the spin-coated thin films. | ||
Quantum chemical calculation
Quantum chemical calculations performed using the B3LYP/6-31G (d,p) method26 reveal that the 2nd generation dendritic dendrimers (G2PYR and G2TPA) adopt more sterically hindering molecular structures than their corresponding 1st generation dendrimers (G1PYR and G1TPA) (Fig. 2). Such structural characteristics could play an important role in reducing an undesirable intermolecular interaction in the solid state, and evolving the glass-forming ability and enhancing the thermal stability of the materials.27 The distributions of π-electrons in the HOMOs of GnPYR and GnTPA are mainly on the 3,6-dipyrenylcarbazole and 3,6-bis[(diphenylamino)phenyl]carbazole moieties, respectively. In the LUMOs of GnPYR, the excited electrons are localized on the electron-rich pyrene peripheries, while in the LUMOs of GnTPA such electrons are located primarily on the central carbazole. Moreover, the TDDFT calculation also reveals that the S1 state (S0 → S1 transition) of GnTPA is dominated by the HOMO → LUMO+1 transition, while the S1 state of GnPYR is dominated by the HOMO → LUMO transition (see ESI (Table S2†)). According to the TDDFT outcomes, it can be deduced that G1PYR and G2PYR, and G1TPA and G2TPA would have similar electronic properties with different physical properties. | ||
| Fig. 2 HOMO and LUMO orbitals of GnPYR and GnTPA calculated by the B3LYP/6-31G(d,p) method in CH2Cl2. | ||
Optical, thermal and electrochemical properties
The UV-vis absorption and photoluminescence PL spectra of GnPYR and GnTPA in CH2Cl2 solution and thin film are shown in Fig. 3 (Table 1). Both G1PYR and G2PYR, and G1TPA and G2TPA exhibit identical solution absorption spectra and optical band gaps (Eoptg), indicating that increasing the size of the carbazole dendritic platforms has no or if any a little effect on the electronic properties of these molecules consistent with the DFT results. GnPYR and GnTPA feature a main absorption peak at 348 and 324 nm attributed to π–π* transitions of the 3,6-dipyrenylcarbazole and 3,6-bis[(diphenylamino)phenyl]carbazole groups, respectively. It is obvious that the absorption spectra of GnPYR exhibit a large red shift of 24 nm compared with those of GnTPA, which could be derived from the extended π conjugation in the 3,6-dipyrenylcarbazole. The PL spectra in solution and thin film of GnPYR and GnTPA show an emission peak in blue region. We found that the PL spectra of GnPYR in thin film are slightly red shifted (9–14 nm) to those in solution, indicating weak intermolecular interactions between GnPYR molecules in the solid state packing. This phenomenon may be caused by the planar structure of the 3,6-dipyrenylcarbazole moiety. With introducing the pyrene fluorophores (GnPYR, ΦF = 0.94–0.95) to the surface of the dendrimers, the fluorescent quantum yield dramatically increased compared with the triphenylamine moieties (GnTPA, ΦF = 0.57–0.61). | ||
| Fig. 3 (a) UV-vis absorption and (b) PL spectra measured in CH2Cl2 (solid line) and as spin-coated thin film (dotted line) on quartz substrates. | ||
| Compd | λabs (logε)a (nm, M−1 cm−1) |
λsolema (nm) | λfilmemb (nm) | Stokes shiftc (nm) | ΦFd | Tg/Tc/Tm/T5de (°C) | E1/2 vs. Ag/AgClf (V) | Eoptg/Ecalgg (eV) | HOMO/LUMOh (eV) |
|---|---|---|---|---|---|---|---|---|---|
| a Measured in CH2Cl2.b Measured as a spin coated thin film.c Calculated from the difference between λmax of absorption and emission spectra in solution.d Measured in CH2Cl2 with quinine sulphate in 0.1 M H2SO4 (ΦF = 0.54) as a reference.e Obtained from DSC/TGA measured at 10 °C min−1 under N2.f Obtained from CV measured in CH2Cl2/n-Bu4NPF6 (0.1 M) at scan rate of 50 m s−1.g Calculated from Eoptg = 1240/λonset; Ecalg calculated by B3LYP/6-31G(d,p) in CH2Cl2 solvent.h Calculated from HOMO = −(4.44 + Eonsetox); LUMO = HOMO + Eoptg. | |||||||||
| G1PYR | 348 (3.80) | 431 | 445 | 83 | 0.94 | 70/113/181/375 | 0.69 (Epc), 0.93 (Epc) 1.09, 1.26, 1.51 | 3.12/3.15 | −5.45/−2.33 |
| G2PYR | 348 (3.88) | 438 | 447 | 90 | 0.95 | 211/—/—/400 | 0.57 (Epc), 1.06 (Epa), 1.66 | 3.12/3.15 | −5.44/−2.32 |
| G1TPA | 324 (5.03) | 415 | 398 | 94 | 0.57 | 52/—/186/370 | 0.79, 0.89, 1.20 | 3.28/3.40 | −5.15/−1.87 |
| G2TPA | 324 (5.14) | 417 | 400 | 90 | 0.61 | 145/—/—/417 | 0.81, 0.90, 1.19 | 3.28/3.31 | −5.17/−1.89 |
The thermal properties of GnPYR and GnTPA were determined by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) (Fig. 4a and Table 1). TGAs reveal that their decomposition temperatures (T5d) at 5 wt% weight loss are above 370 °C, showing the high thermal stability. DSCs (1st and 2nd heating scans) of the 2nd generation dendrimers, G2PYR and G2TPA, reveal a distinct glass transition temperature (Tg) at 211 and 145 °C, respectively, and no crystallization and melting peaks being observed at higher temperatures, indicating an excellent amorphous glass state stability.31 DSC (1st heating scan) of G1PYR having smaller scaffold shows an endothermic baseline shift at 70 °C (Tg) followed by exothermic crystallization and endothermic melting peaks at 113 and 181 °C, respectively, while the DSC (1st heating scan) of G1TPA exhibits only a distinct melting peak at 186 °C. The subsequent DSC scan of G1PYR and G1TPA show only a Tg at 70 and 52 °C, respectively. The high Tgs of G2PYR and G2TPA may be attributed to the more bulky rigid structure of the 2nd generation carbazole dendritic skeleton. Operating temperatures of the OLEDs exceeding the Tg of the active organic materials are likely to promote thermally activated degradation processes and will induce device failure. Devices incorporating amorphous thin films having high Tg are less vulnerable to heat. Organic materials with high Tg are therefore highly desirable for applications in long lifetime electroluminescent devices.28
 | ||
| Fig. 4 (a) DSC (1st heating (solid line) and 2nd heating (dotted line) scans) and TGA thermograms measured at a heating rate of 10 °C min−1 under N2 flow. (b) CV traces of GnPYR and GnTPA and repeated CV scans of (c) G2TPA and (d) G2PYR of measured in CH2Cl2/n-Bu4NPF6 at a scan rate of 50 mV s−1 under argon flow. | ||
The redox behaviours of GnPYR and GnTPA were investigated by means of cyclic voltammetry measurements (Fig. 4b–d and Table 1). CVs of GnTPA are nearly the same and display three quasi-reversible oxidation processes at E1/2 of 0.8, 0.9 and 1.2 V. The first oxidation process assigns to the removal of electrons from the peripheral triphenylamine, resulting in radical cations. Vitally, the repeated CV scans of GnTPA reveal identical CV traces, suggesting they are electrochemically stable molecules. Under the same CV measurement conditions, on the contrary, GnPYR exhibit a series of irreversible oxidation processes and an additional peak at a lower potential on the cathodic scan (Epc) around 0.57–0.69 V. Their repeated CV scans display an increasing change in the CV traces, signifying that a series of electrochemical reactions led to electro-polymerization of the radical cation species occurring on the glassy carbon electrode surface (Fig. S2†).6 However, this type of radical–radical coupling reaction will become inactive in a non-diffusion system or in the device. In addition, under these measurement conditions, no reduction process is observed in all cases. The energy levels of the HOMO and LUMO of these compounds were determined and are listed in Table 1. Their HOMO levels, estimated from CV and optical results, are in the range of −5.15 to −5.45 eV, which are close to the work function of a commonly used indium tin oxide (ITO) anode (−4.80 eV). Their LUMO levels range from −1.87 eV to −2.33 eV, which are close to the work function of LiF/Al cathode (−2.60 eV).
Electroluminescence (EL) properties
:toluene (1:1) solution. Conductive poly(3,4-ethylenedioxythiophene):poly(4-styrenesulfonate) (PEDOT:PSS) as hole injection layer and 2,9-dimethyl-4,7-diphenyl-1,10-phenanthroline (BCP) as hole blocking layer were incorporated to enable high efficiency diodes.29 The electroluminescence data of the devices are shown in Fig. 6 and 7 and listed in Table 2. Under a bias voltage, all devices (I–III) emit a strong blue emission with λEL peaked at 437, 463 and 430 nm and a narrow FWHM of 67–90 nm, respectively (Fig. 7a). The electroluminescence (EL) spectra of devices I and II match with their corresponding PL spectra, indicating that the EL purely originates from GnPYR layers. Both devices I–II show a pure blue emission with CIE color coordinates of (0.15, 0.13) and (0.15, 0.16), respectively. Upon varying the bias voltages, no emission shoulder at the longer wavelength and no significant change in the EL spectra of devices I and II causing by the emission of the excimer and exciplex species formed at the EML/BCP interface, which often occurs in the devices fabricated from EML with planar molecular structure, is noticed.30 In our case, the formation of such species could be prohibited by the bulky nature of the carbazole dendrimer implemented as molecular platform. As depicted in Fig. 7, GnPYR-based blue OLEDs (devices I and II) show superior luminance efficiency than that of the reference NPB-based blue device (device III). The light turn-on voltage (Von) at 1 cd m−2 of these devices is 3.1 V and the operating voltage (V100) at 100 cd m−2 is in the range of 4.4–5.1 V, signifying a decent device performance. G2PYR-based diode (device II) exhibits the best device performance with a maximum brightness (Lmax) of 23003 cd m−2, a maximum luminance efficiency (ηmax) of 5.89 cd A−1, a maximum power efficiency (PE) of 2.67 lm W−1 and a high external quantum efficiency (EQE) of 6.81%. G1PYR-based diode (device I) shows a slightly lower device performance with an Lmax of 11964 cd m−2, a ηmax of 3.67 cd A−1, a PE of 1.68 lm W−1 and an EQE of 5.11%. The higher EL efficiency of the G2PYR-based device than G1PYR-based device may stem from a combination of a better thin film-forming quality and stability of G2PYR.31
 | ||
| Fig. 5 Schematic energy diagrams of the OLEDs fabricated with (a) GnPYR as EML and (b) with GnPYR and GnTPA as HTL. (c) Molecular structures of NPB, Alq3 and BCP. | ||
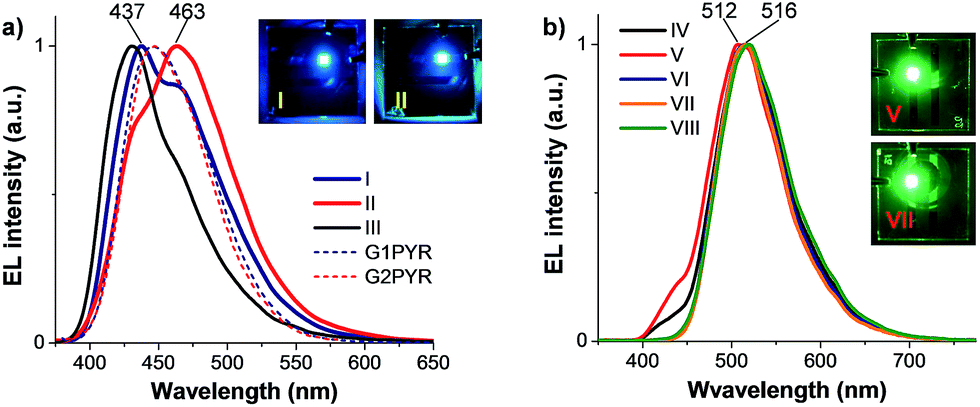 | ||
| Fig. 6 Thin film PL spectra (dotted line) of the EML and EL spectra (solid line) of the OLEDs fabricated with (a) GnPYR as EML and (b) with GnPYR and GnTPA as HTL, and their emission colours under applied voltages. | ||
 | ||
| Fig. 7 Plots of (a) and (c) current density–luminance–voltage (J–V–L), and (b) and (d) efficiency–current density (η–J) characteristics of the OLEDs. | ||
| Device | EML/HTL | λELmax/FWHM (nm) | Von/V100c (V) | Lmax at voltaged (cd m−2/V) | Jmaxe (mA cm−2) | ηmax/η at L100/η at L1000f (cd A−1) | EQEg (%) | PE at voltageh (lm W−1 V−1) | CIE (x, y) |
|---|---|---|---|---|---|---|---|---|---|
| a ITO/PEDOT:PSS/EML/BCP/LiF:Al.b ITO/PEDOT:PSS/HTL/Alq3/LiF:Al.c Turn-on voltages at 1 and 100 cd m−2.d Maximum luminance at applied voltage.e Current density.f Luminance efficiencies at maximum, at luminance of 100 and 1000 cd m−2.g Maximum external quantum efficiency.h Maximum power efficiency at applied voltage. | |||||||||
| Ia | G1PYR | 437, 463sh/83 | 3.1/5.1 | 11964 (10.8) |
533 | 3.67/2.47/3.55 | 5.11 | 1.68 (6.2) | 0.15, 0.13 |
| IIa | G2PYR | 433sh, 463/90 | 3.1/4.4 | 23003 (11.4) |
607 | 5.89/3.00/5.22 | 6.81 | 2.67 (6.8) | 0.15, 0.16 |
| IIIa | NPB | 430/67 | 3.0/4.1 | 7189 (9.0) | 822 | 1.95/1.85/1.90 | 2.72 | 1.61 (3.2) | 0.16, 0.09 |
| IVb | G1PYR | 517/82 | 2.6/3.4 | 31201 (10.0) |
1057 | 4.48/3.65/4.30 | 1.11 | 3.37 (3.4) | 0.27, 0.50 |
| Vb | G2PYR | 512/82 | 2.6/3.4 | 33846 (9.8) |
1209 | 5.15/4.20/4.94 | 1.28 | 3.84 (3.5) | 0.25, 0.46 |
| VIb | G1TPA | 515/83 | 2.7/3.7 | 23580 (10.6) |
939 | 4.79/4.40/4.70 | 1.19 | 3.86 (3.4) | 0.27, 0.53 |
| VIIb | G2TPA | 516/82 | 2.7/3.7 | 27787 (10.6) |
764 | 6.10/5.40/5.92 | 1.51 | 4.64 (3.4) | 027, 0.54 |
| VIIIb | NPB | 519/83 | 2.8/3.8 | 35631 (12.0) |
1670 | 4.89/4.33/4.75 | 1.21 | 3.72 (3.6) | 0.29, 0.53 |
787–33846 cd m−2 for green OLED at 9.8–10.6 V, low Von of 2.6–2.7 V, ηmax of 5.15–6.10 cd A−1, PE of 3.84–4.64 lm W−1 and EQE of 1.28–1.51%. The operating voltage at 100 cd m−2 of these diodes is as low as 3.4 V. The green OLEDs (devices IV and VI) fabricated with G1PYR and G1TPA as HTL show a slightly lower device performance with ηmax of 4.48–4.79 cd A−1, PE of 3.37–3.86 lm W−1 and EQE of 1.11–1.19%. Better film forming ability and quality of G2PYR and G2TPA may be the key to their superior hole-transport property and performance to those of devices using G1PYR and G1TPA. By comparison the performance of both devices VII and V with the reference NPB-based device VIII, the integration of either G2PYR or G2TPA in the device as HTL not only increases the ηmax from to 4.89 cd A−1 to 5.15–6.10 cd A−1, but also decreases the Von from 2.8 to 2.6 V, indicating an excellent solution-processed HTM ability of both materials. Besides, the ability as amorphous HTM of G2PYR and G2TPA in terms of thermal stability of amorphous films (Tg = 145–211 °C) is also superior than commonly used NPB (Tg = 100 °C).Although the luminance efficiency of G2PYR-based OLEDs as both EML and HTL cannot compete with reported iridium complex-based phosphorescent OLEDs (ηmax = 53.5 cd A−1 and EQE = 20.1%)33 and some thermally activated delayed fluorescence (TADF) OLEDs (EQE = 14.5%),34 surely the advantage of the blue and green OLEDs in this study is a simple device fabrication process and structure. Wang et al. have reported pyrene derivatives as blue-light emitting and hole-transporting bifunctional materials. They exhibit a luminance efficiency of 0.97 cd A−1 with wavelength at 440 nm when used as a blue emitter and a luminance efficiency of 0.40 cd A−1 with wavelength at 453 nm when used as a hole-transporting material.35 verifying that the ability of G2PYR as a non-doped hole-transporting blue emitter, in terms of device performance, is excellence. The performance of G2PYR is also better than our recent reported anthracene- and oligofluorene-cored carbazole dendrimer-based bifunctional materials.36
Experimental
Materials and methods
All starting materials were obtained from commercial suppliers and used without further purification; solvents were purified according to standard techniques. Compound 1 was synthesized following the reported procedure.371H and 13C NMR spectra were recorded on an AVANCE 300 MHz spectrometer. UV-Vis spectra were recorded on an UV Lambda 25 spectrometer. Photoluminescence spectra and fluorescence quantum yields (ΦF) were measured with a LS 50B Luminescence spectrometer. Differential scanning calorimetry (DSC) and thermal gravimetric analysis (TGA) were performed on a DSC823e and TG-DTA 8120 thermal analyzers, respectively, with heating rate of 10 °C min−1 under N2 flow. Cyclic voltammetry (CV) was carried out on a PGSTAT 12 with a three electrode system (platinum counter electrode, glassy carbon working electrode and Ag/AgCl reference electrode) at scan rate of 50 mV s−1 in the presence of n-Bu4NPF6 as a supporting electrolyte in CH2Cl2 under argon flow. Melting points were measured using an Electrothermal IA 9100 series of digital melting point instrument and are uncorrected. High-resolution mass spectrometry (HRMS) analysis was performed on an Autoflex II MALDI-TOF/TOF mass spectrometer. The atomic force microscopy (AFM) analysis was performed on an XE 100.
All calculations were performed by Gaussian 09 code in CH2Cl2 solvent.26 The energy or geometry optimizations were done by B3LYP/6-31G(d,p) method. The ground to excited state excitation energies were calculated by B3LYP/6-31G(d,p) in CH2Cl2.
Materials synthesis
:4) followed by recrystallization with a mixture of CH2Cl2 and CH3OH afforded the product (2.15 g, 80%) as colorless solids: mp 232 °C; 1H NMR (300 MHz, CDCl3) δ 8.26 (2H, s), 8.21 (4H, d, J = 7.80 Hz), 7.68 (4H, s), 7.47–7.41 (8H, m), 7.36–7.28 (4H, m), 4.49 (2H, t, J = 7.20 Hz), 2.06 (2H, t, J = 6.90 Hz), 1.56–1.33 (18H, m), 0.93 (3H, t, J = 6.30 Hz) ppm; 13C NMR (300 MHz, CDCl3) δ 141.91, 129.35, 125.98, 125.87, 123.15, 120.29, 119.86, 110.13, 109.77, 43.15, 31.94, 29.67, 29.61, 29.51, 29.38, 29.21, 27.46, 22.86, 14.29 ppm; MALDI-TOF (m/z) (M+) calcd for C48H47N3: 665.9069, found 665.4128.:4) followed by recrystallization with a mixture of CH2Cl2 and CH3OH gave the product (1.43 g, 98%) as white solids: mp 150 °C; 1H NMR (300 MHz, CDCl3) δ 8.21 (4H, s), 8.16 (2H, s), 7.68 (2H, d, J = 8.70 Hz), 7.57 (2H, d, J = 8.70 Hz), 7.48 (4H, d, J = 8.55 Hz), 7.22 (4H, d, J = 8.70 Hz), 4.49 (2H, t, J = 7.20 Hz), 2.04 (2H, t, J = 6.90 Hz), 1.57–1.27 (18H, m), 0.87 (3H, t, J = 6.30 Hz) ppm; 13C NMR (300 MHz, CDCl3) δ 140.82, 140.45, 129.33, 128.46, 126.61, 125.78, 123.32, 123.21, 119.72, 112.85, 111.48, 110.45, 43.78, 31.92, 29.64, 29.58, 29.46, 29.35, 29.17, 27.41, 22.69, 14.12 ppm; MALDI-TOF (m/z) (M+) calcd for C48H43Br4N3: 977.0190, found 977.1070.Synthesis of GnPYR and GnTPA
A mixture of 1 or 3 (0.61 mmol), pyrene-1-boronic acid or 4-(diphenylamino)phenylboronic acid (1.40–3.10 mmol), Pd(PPh3)4 (0.02 mmol) and 2 M Na2CO3 solution (7 ml) in THF (25 ml) was degassed with N2 for 5 min. The mixture was refluxed under N2 for 48 h. After cooling, water (30 ml) was added and the mixture was extracted with CH2Cl2 (50 ml × 2). The combined organic layer was washed with water (50 ml), brine solution (50 ml), dried over anhydrous Na2SO4, filtered and removed to dryness. Purification by column chromatography over silica gel eluting with a mixture of CH2Cl2 and hexane (1:4) followed by recrystallization with a mixture of CH2Cl2 and CH3OH yielded:
G1PYR as light green solids (90%): mp 200 °C; 1H NMR (300 MHz, CDCl3) δ 8.41 (2H, s), 8.34 (2H, d, J = 9.30 Hz), 8.25 (2H, d, J = 7.80 Hz), 8.20–8.10 (10H, m), 8.06–7.97 (4H, m), 7.79 (2H, d, J = 8.40 Hz), 7.66 (2H, d, J = 7.64 Hz), 4.47 (2H, t, J = 7.20 Hz), 2.06 (2H, q, J = 6.90 Hz), 1.57–1.30 (18H, m), 0.91 (3H, t, J = 6.90 Hz) ppm; 13C NMR (300 MHz, CDCl3) δ 140.31, 138.68, 132.10, 131.58, 131.10, 130.29, 128.89, 128.76, 128.25, 127.51, 127.32, 127.17, 125.93, 125.74, 125.11, 125.04, 124.93, 124.66, 123.11, 122.48, 108.71, 43.55, 31.96, 30.93, 29.69, 29.55, 29.40, 29.25, 27.50, 22.73, 14.16 ppm; MALDI-TOF (m/z) (M+) calcd for C56H49N: 735.3865, found 735.2479.
G2PYR as light green solids (91%): mp > 250 °C; 1H NMR (300 MHz, CDCl3) δ 8.49 (6H, s), 8.33 (4H, d, J = 9.3 Hz), 8.22 (4H, d, J = 7.8 Hz), 8.19–8.05 (20H, m), 8.02 (4H, d, J = 3.0 Hz), 7.98 (4H, d, J = 7.8 Hz), 7.88 (2H, d, J = 8.4 Hz), 7.80–7.74 (6H, m), 7.68 (4H, d, J = 8.4 Hz), 4.55 (2H, bs), 2.11 (2H, bs), 1.48 (2H, bs), 1.27 (16H, bs), 0.86 (3H, t, J = 6.6 Hz) ppm; 13C NMR (300 MHz, CDCl3) δ 141.80, 140.45, 138.57, 133.09, 131.55, 131.06, 130.34, 129.40, 129.07, 128.89, 128.20, 127.48, 127.37, 127.19, 126.05, 125.92, 125.68, 125.07, 125.00, 124.94, 124.68, 124.64, 123.62, 123.46, 122.42, 119.97, 110.39, 109.84, 43.83, 31.92, 30.92, 29.66, 29.63, 29.53, 29.36, 29.26, 27.49, 22.69, 14.11 ppm; MALDI-TOF (m/z) (M+) calcd for C112H79N3: 1465.6274, found 1465.5541.
G1TPA as white solids (89%): mp 200 °C; 1H NMR (300 MHz, CDCl3) δ 8.36 (2H, s), 7.73 (2H, dd, J = 8.40 Hz, J = 1.2 Hz), 7.64 (2H, d, J = 8.40 Hz), 7.47 (2H, d, J = 8.40 Hz), 7.33–7.28 (8H, m), 7.24–7.18 (12H, m), 7.07 (4H, t, J = 7.20 Hz), 4.34 (2H, t, J = 7.20 Hz), 1.93 (2H, q, J = 6.90 Hz), 1.52–1.29 (18H, m), 0.90 (3H, t, J = 6.90 Hz) ppm; 13C NMR (300 MHz, CDCl3) δ 147.92, 146.43, 140.24, 136.51, 131.94, 129.27, 127.92, 125.10, 124.99, 124.54, 124.22, 123.56, 122.71, 118.48, 109.06, 43.37, 31.95, 29.74, 29.55, 29.46, 29.36, 29.10, 27.37, 22.72, 14.15 ppm; MALDI-TOF (m/z) (M+) calcd for C60H59N3: 821.4709, found 821.1257.
G2TPA as white solids (85%). mp > 250 °C; 1H NMR (300 MHz, CDCl3) δ 8.39 (4H, s), 8.30 (2H, s), 7.72 (6H, s), 7.65–7.60 (12H, m), 7.45 (6H, d, J = 8.40 Hz), 7.30–7.25 (16H, m), 7.20–7.15 (20H, m), 7.03 (8H, t, J = 7.20 Hz), 4.52 (2H, t, J = 7.20 Hz), 2.16–2.06 (2H, m), 1.61–1.26 (18H, m), 0.89 (3H, t, J = 6.90 Hz) ppm; 13C NMR (300 MHz, CDCl3) δ 147.85, 146.49, 141.58, 140.25, 136.37, 132.88, 130.87, 129.36, 123.223, 128.81, 127.93, 125.84, 125.26, 124.45, 124.20, 123.83, 123.45, 122.69, 119.70, 118.39, 110.10, 43.79, 38.76, 31.92, 30.90, 30.39, 29.70, 29.65, 29.36, 28.94, 27.45, 23.77, 22.98, 22.69, 14.11 ppm; MALDI-TOF (m/z) (M+) calcd for C120H99N7: 1637.7962, found 1637.4813.
OLED fabrication and testing
All OLED devices using GnPYR and GnTPA as hole-transporting layer (THL) and GnPYR as blue emissive layer (EML) with the device structures of ITO/PEDOT:PSS/THL(spin-coating) (30–40 nm)/Alq3(50 nm)/LiF(0.5 nm):Al(150 nm) and ITO/PEDOT:PSS/EML(spin-coating) (30–40 nm)/BCP(40 nm)/LiF(0.5 nm):Al(150 nm) were fabricated and characterized as followed. Thin films of GnPYR and GnTPA were deposited on top of PEDOT:PSS coated ITO by spin-coating CHCl3:toluene solutions (1:1) of GnPYR and GnTPA (1.0–2.0% w/v) at a spin speed of 3000 rpm for 30 second to get a 30–40 nm thick of THL/EML. The film thickness was measured by using a Tencor α-Step 500 surface profiler. The BCP/Alq3 layer was deposited on top as electron-transporting layer (ETL)/EML with a thickness of 40–50 nm by evaporation from resistively heated alumina crucible at evaporation rate of 0.5–1.0 nm s−1 in vacuum evaporator deposition (ES280, ANS Technology) under a base pressure of ∼10−5 mbar. The film thickness was monitored and recorded by quartz oscillator thickness meter (TM-350, MAXTEK). A 0.5 nm thick LiF and a 150 nm thick Al layers were the subsequently deposited through a shadow mask on the top of BCP/Alq3 film without braking vacuum to from an active diode areas of 4 mm2. The measurement of device efficiency was performed according to M. E. Thomson's protocol and the device power efficiencies were calculated using procedure reported previously.38 Current density–voltage–luminescence (J–V–L) characteristics were measured simultaneous by the use of a Keithley 2400 source meter and a Newport 1835C power meter equipped with a Newport 818-UV/CM calibrated silicon photodiode. The EL spectra were recorded by an Ocean Optics USB4000 multichannel spectrometer. All the measurements were performed under ambient atmosphere at room temperature soon after breaking the chamber.
Conclusions
In summary, we have successfully synthesized a series of pyrene- and triphenylamine-functionalized carbazole dendrimers as bifunctional materials for OLEDs. By using carbazole dendrimers (1st and 2nd generations) as a scaffold, we are able to retain the high emissive ability and reduce the excimer emission of pyrene fluorophore in the solid state as well as improve hole-transporting property, amorphous glass-forming ability, solution processability and thermal stability of the material. Particularly, pyrene- and triphenylamine-functionalized 2nd generation carbazole dendrimers, G2PYR and G2TPA, exhibit a strong blue emission with morphologically and thermally stable amorphous thin films (Tg = 145–211 °C). A solution processed blue OLED (ITO/PEDOT:PSS/G2PYR(spin coating)/BCP/LiF–Al) using G2PYR as an emissive layer shows an excellent luminance efficiency of 5.89 cd A−1 (EQE = 6.81%) and a pure blue emission with CIE coordinates of (0.15, 0.16), while Alq3-based green OLED (ITO/PEDOT:PSS/G2PYR(spin coating)/Alq3/LiF–Al) using G2PYR as hole-transporting layer displays a high luminance efficiency of 6.10 cd A−1 (EQE = 1.51%). These results revealed that G2PYR is promising as blue emitter and hole-transporter for high efficiency OLEDs with much simpler device architecture. We believe that our results could provide a useful guidance to decorate the highly efficient but planar fluorophore to be suitable for applications in solution-processable and non-doped OLEDs without a hole-transporting layer.Acknowledgements
This work was supported by the Thailand Research Fund (TRF) (Grant No. DBG5580001 and RTA5680008) and Rayong Institute of Science and Technology (RAIST) Foundation. We thank Department of Physics, Ubon Ratchathani University for providing AFM facility.Notes and references
- C. H. Chen, J. Shi and C. W. Tang, Macromol. Symp., 1997, 125, 1 CrossRef PubMed; B. W. D'Andrade and S. R. Forrest, Adv. Mater., 2004, 16, 1585 CrossRef PubMed.
- N. Matsumoto, T. Miyazaki, M. Nishiyama and C. Adachi, J. Phys. Chem. C, 2009, 113, 6261 Search PubMed; X. Zhan, Y. Liu, X. Wu, S. Wang and D. Zhu, Macromolecules, 2002, 35, 2529 CrossRef CAS; T. Spehr, R. Pudzich, T. Fuhrmann and J. Salbeck, Org. Electron., 2003, 4, 61 CrossRef PubMed; C. C. Wu, Y. T. Lin, H. H. Chiang, T. Y. Cho, C. W. Chen, K. T. Wong, Y. L. Liao, G. H. Lee and S. M. Peng, Appl. Phys. Lett., 2002, 81, 577 CrossRef PubMed.
- C. H. Chen and J. M. Shi, Coord. Chem. Rev., 1998, 171, 161 CrossRef CAS.
- Y. Hamada, IEEE Trans. Electron Devices, 1997, 44, 1208 CrossRef CAS.
- F. He, H. Xu, B. Yang, Y. Duan, L. Tian, K. Huang, Y. Ma, S. Liu, S. Feng and J. Shen, Adv. Mater., 2005, 17, 2710 CrossRef CAS PubMed; C. Hosokawa, H. Higashi, H. Nakamura and T. Kusumoto, Appl. Phys. Lett., 1995, 67, 3853 CrossRef PubMed.
- S. L. Lai, Q. X. Tong, M. Y. Chan, T. W. Ng, M. F. Lo, C. C. Ko, S. T. Lee and C. S. Lee, Org. Electron., 2011, 12, 541 CrossRef CAS; B. Wang, J. Zhao, J. Xiao, C. Cui and R. Liu, Int. J. Electrochem. Sci., 2012, 7, 2781 Search PubMed; T. M. Figueira-Duarte and K. Müllen, Chem. Rev., 2011, 111, 7260 CrossRef PubMed; T. Kumchoo, V. Promarak, T. Sudyoadsuk, M. Sukwattanasinitt and P. Rashatasakhon, Chem.–Asian J., 2010, 5, 2162 CrossRef PubMed; J. Hu and T. Yamato, Organic Light Emitting Diode-Material, Process and Devices, InTech, 2011, pp. 21–60 Search PubMed.
- K. Danel, T. Huang, J. T. Lin, Y. Tao and C. Chuen, Chem. Mater., 2002, 14, 3860 CrossRef CAS; N. Prachumrak, S. Namuangruk, T. Keawin, S. Jungsuttingwong, T. Sudyoadsuk and V. Promarak, Eur. J. Org. Chem., 2013, 18, 3825 CrossRef PubMed.
- Y. Sato, S. Ichinosawa, T. Ogata, M. Fugono and Y. Murata, Synth. Met., 2000, 111–112, 25 CrossRef CAS.
- E. Balasubramaniam, Y. T. Tao, A. Danel and P. Tomasik, Chem. Mater., 2000, 12, 2788 CrossRef CAS.
- K.-C. Wu, P.-J. Ku, C.-S. Lin, H.-T. Shih, F.-I. Wu, M.-J. Huang, J.-J. Lin, I.-C. Chen and C.-H. Cheng, Adv. Funct. Mater., 2008, 18, 67 CrossRef CAS PubMed; F. Liu, C. Tang, Q.-Q. Chen, F.-F. Shi, H.-B. Wu, L.-H. Xie, B. Peng, Y. Cao and W. Huang, J. Phys. Chem. C, 2009, 113, 4641 Search PubMed.
- F. Wurthner, C. Thalacker, S. Dieke and C. Tschierske, Chem.–Eur. J., 2001, 7, 2245 CrossRef CAS; S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765 Search PubMed.
- T. Keawin, C. Sooksai, N. Prachumrak, T. Kaewpuang, D. Muenmart, S. Namuangruk, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, RSC Adv., 2015, 5, 16422 RSC.
- J. K. Salunke, P. Sonar, F. L. Wong, V. A. L. Roy, C. S. Lee and P. P. Wadgaonkar, Phys. Chem. Chem. Phys., 2014, 16, 23320 RSC; P. Sonar, M. S. Soh, Y. H. Cheng, J. T. Henssler and A. Sellinger, Org. Lett., 2010, 12, 3292 CrossRef CAS PubMed; J. Narasimha Moorthy, P. Natarajan, P. Venkatakrishnan, D.-F. Huang and T. J. Chow, Org. Lett., 2007, 9, 5215 CrossRef PubMed.
- S. Tao, Y. Zhou, C.-S. Lee, X. Zhang and S.-T. Lee, Chem. Mater., 2010, 22, 2138 CrossRef CAS.
- K. L. Chan, J. P. F. Lim, X. Yang, A. Dodabalapur, G. E. Jabbour and A. Sellinger, Chem. Commun., 2012, 48, 5106 RSC; X. H. Yang, T. Giovenzana, B. Feild, G. E. Jabbour and A. Sellinger, J. Mater. Chem., 2012, 22, 12689 RSC.
- C. Tang, F. Liu, Y.-J. Xia, J. Lin, L.-H. Xie, G.-Y. Zhong, Q.-L. Fan and W. Huang, Org. Electron., 2006, 7, 155 CrossRef CAS PubMed.
- A. Thangthong, N. Prachumrak, R. Tarsang, T. Keawin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, J. Mater. Chem., 2012, 22, 6869 RSC.
- Z. Zhao, X. Xu, H. Wang, P. Lu, G. Yu and Y. Liu, J. Org. Chem., 2008, 73, 594 CrossRef CAS PubMed.
- M. Y. Lo, C. Zhen, M. Lauters, G. E. Jabbour and A. Sellinger, J. Am. Chem. Soc., 2007, 129, 5808 CrossRef CAS PubMed.
- X.-H. Zhu, J. Peng, Y. Cao and J. Roncali, Chem. Soc. Rev., 2011, 40, 3509 RSC; L. Duan, L. Hou, T.-W. Lee, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. Qiu, J. Mater. Chem., 2010, 20, 6392 RSC.
- J. Li, Q. Li and D. Liu, ACS Appl. Mater. Interfaces, 2011, 3, 2099 Search PubMed; T. Sudyoadsuk, P. Moonsin, N. Prachumrak, S. Namuangruk, S. Jungsuttiwong, T. Keawin and V. Promarak, Polym. Chem., 2014, 5, 3982 RSC; C. Liu, Q. Fu, Y. Zou, C. Yang, D. Ma and J. Qin, Chem. Mater., 2014, 26, 3074 CrossRef CAS; S.-C. Lo, R. E. Harding, C. P. Shipley, S. G. Stevenson, P. L. Burn and I. D. W. Samuel, J. Am. Chem. Soc., 2009, 131, 16681 CrossRef PubMed; Y. Li, A.-Y. Li, B.-X. Li, J. Huang, L. Zhao, B.-Z. Wang, J.-W. Li, X.-H. Zhu, J. Peng, Y. Cao, D.-G. Ma and J. Roncali, Org. Lett., 2009, 11, 5318 CrossRef PubMed; S.-H. Hwang, C. N. Moorefield and G. R. Newkome, Chem. Soc. Rev., 2008, 37, 2543 RSC.
- W. L. Jia, D. R. Bai, T. McCormick, Q. D. Liu, M. Motala, R. Y. Wang, C. Seward, Y. Tao and S. Wang, Chem.–Eur. J., 2004, 4, 994 CrossRef CAS PubMed; P. C. Bevilacqua, R. Kierzek, K. A. Johnson and D. H. Turner, Science, 1992, 258, 1355 Search PubMed.
- P. Moonsin, N. Prachumrak, R. Rattanawan, T. Keawin, S. Jungsuttiwong, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2012, 48, 3382 RSC.
- P. Thongkasee, A. Thangthong, N. Janthasing, T. Sudyoadsuk, S. Namuangruk, T. Keawin, S. Jungsuttiwong and V. Promarak, ACS Appl. Mater. Interfaces, 2014, 6, 8212 CAS.
- R. Schmechel and H. von Seggern, Phys. Status Solidi A, 2004, 201, 1215 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1, Gaussian Inc., Wallingford CT, 2009 Search PubMed.
- Z. J. Ning, Y. C. Zhou, Q. Zhang, D. G. Ma, J. J. Zhang and H. Tian, J. Photochem. Photobiol., A, 2007, 192, 8 CrossRef CAS PubMed.
- P. Strohriegl and J. V. Grazulevicius, Adv. Mater., 2002, 14, 1439 CrossRef CAS; R. Fink, Y. Heischkel, M. Thelakkat, H.-W. Schmidt, C. Jonda and M. Hüppauff, Chem. Mater., 1998, 10, 3620 CrossRef.
- W. Kim, G. Kushto, G. Kim and Z. Kafafi, J. Polym. Sci., Part B: Polym. Phys., 2003, 41, 2522 CrossRef CAS PubMed; S. T. Zhang, Z. J. Wang, J. M. Zhao, Y. Q. Zhan, Y. Wu, Y. C. Zhou, X. M. Ding and X. Y. Houa, Appl. Phys. Lett., 2004, 84, 2916 CrossRef PubMed.
- D. Y. Kim, H. N. Cho and C. Y. Kim, Prog. Polym. Sci., 2000, 25, 1089 CrossRef CAS.
- U. Mitschke and P. Bäuerle, J. Mater. Chem., 2000, 10, 1471 RSC.
- Y. K. Kim and S.-H. Hwang, Synth. Met., 2006, 156, 1028 CrossRef CAS PubMed; C.-H. Chen, W.-J. Shen, K. Jakka and C.-F. Shu, Synth. Met., 2004, 143, 215 CrossRef PubMed; A. M. Thaengthong, S. Saengsuwan, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Tetrahedron Lett., 2011, 37, 4749 CrossRef PubMed.
- J. Zhuang, W. Li, W. Su, Y. Liu, Q. Shen, L. Liao and M. Zhou, Org. Electron., 2013, 14, 2596 CrossRef CAS PubMed; F. Kessler, Y. Watanabe, H. Sasabe, H. Katagiri, M. K. Nazeeruddin, M. Grätzel and J. Kido, J. Mater. Chem. C, 2013, 1, 1070 RSC.
- S. Wu, M. Aonuma, Q. Zhang, S. Huang, T. Nakagawa, K. Kuwabara and C. Adachi, J. Mater. Chem. C, 2014, 2, 421 RSC.
- W.-L. Jia, T. M. Cormick, Q.-D. Liu, H. Fukutani, M. Motala, R.-Y. Wang, Y. Tao and S. Wang, J. Mater. Chem., 2004, 14, 334 Search PubMed.
- A. M. Thangthong, D. Meunmart, N. Prachumrak, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, Chem. Commun., 2011, 47, 7122 RSC; P. Moonsin, N. Prachumrak, S. Namuangruk, S. Jungsuttiwong, T. Keawin, T. Sudyoadsuk and V. Promarak, J. Mater. Chem. C, 2014, 2, 5540 RSC.
- T. Sudyoadsuk, S. Pansay, S. Morada, R. Rattanawan, S. Namuangruk, T. Kaewin, S. Jungsuttiwong and V. Promarak, Eur. J. Org. Chem., 2013, 23, 5051 CrossRef PubMed.
- S. R. Forrest, D. D. C. Bradley and M. E. Thomson, Adv. Mater., 2003, 15, 1043 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Quantum chemical calculation data, CV plot, EL spectra and NMR spectra. See DOI: 10.1039/c5ra07161e |
| This journal is © The Royal Society of Chemistry 2015 |