
Nanonickel catalyst reinforced with silicate for methane decomposition to produce hydrogen and nanocarbon: synthesis by co-precipitation cum modified Stöber method†
Abstract
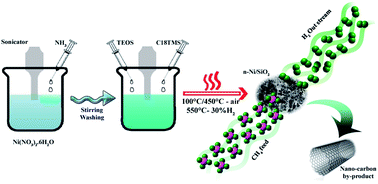
Co-precipitation cum modified Stöber method is a continuous process avoiding application of higher temperature treatment before supporting nanometal with SiO2, irrespective of pre-preparation methods. We have conducted the co-precipitation process without undertaking calcination under air in order to avoid even a partial particle agglomeration and hence maintained average particle size ∼30 nm after enforcing with SiO2. This is the first report adopting such an unceasing preparation for preparing metal/silicate nanostructures. Furthermore, n-Ni/SiO2 nanostructured catalysts were used for thermocatalytic decomposition of methane to produce hydrogen and carbon nanotubes. The catalyst was found to be very stable and the methane transformation activity proceeded for 300 min on methane stream with little deactivation in the temperature range 475–600 °C. We have also successfully extended the catalyst preparation method for Fe and Co metals and conducted preliminary catalyst examinations.
 Please wait while we load your content...
Please wait while we load your content...

