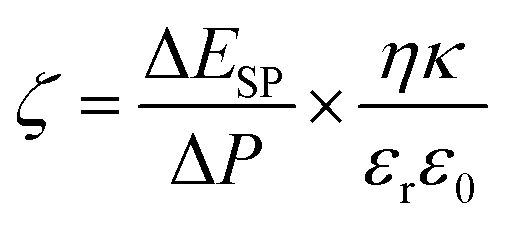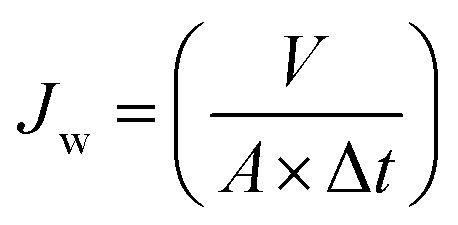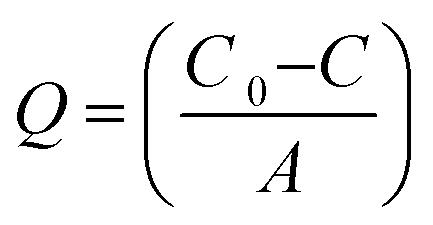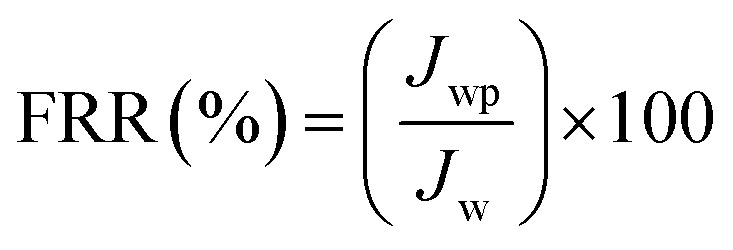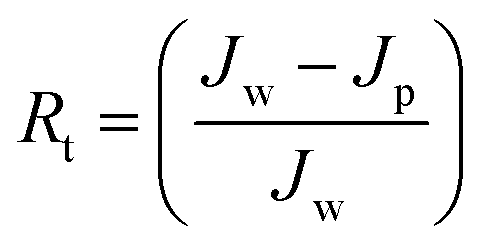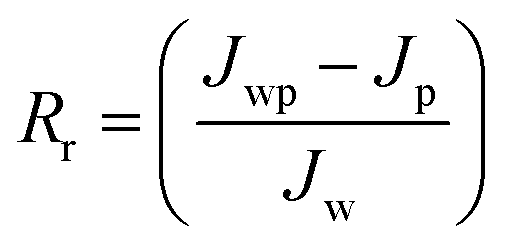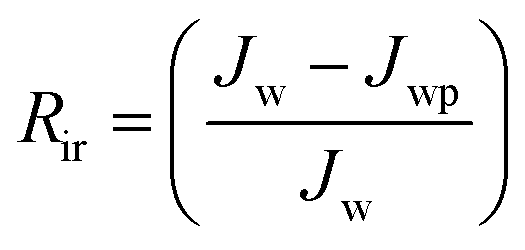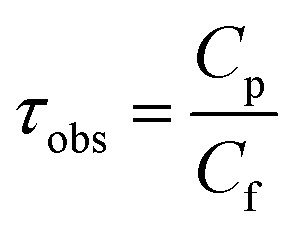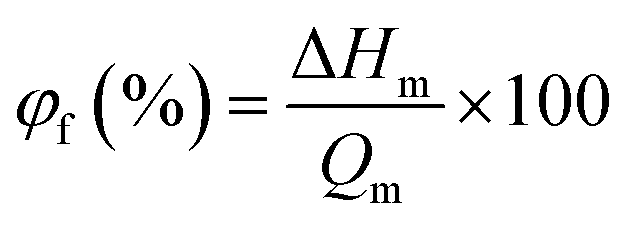DOI:
10.1039/C5RA06893B
(Paper)
RSC Adv., 2015,
5, 51208-51219
Quaternized polysulfone and graphene oxide nanosheet derived low fouling novel positively charged hybrid ultrafiltration membranes for protein separation†
Received
16th April 2015
, Accepted 2nd June 2015
First published on 8th June 2015
Abstract
Low fouling novel positively charged hybrid ultrafiltration membranes with adjustable charge density were fabricated from blends of polysulfone (PSf) and quaternized polysulfone (QPSf) in combination with varied fractions of graphene oxide (GO) nanosheets by a non-solvent induced phase separation method. Fourier transform infrared spectroscopy in the attenuated total reflection mode, scanning electron microscopy, outer surface zeta potential and contact angle studies were conducted to characterize the morphologies of hybrid membranes, structures, charge and surface properties. Results confirmed the fabrication of porous, hydrophilic and positively charged membranes. The water permeabilities (flux) and antifouling ability of membranes with protein solution were dependent on the fraction of quaternary ammonium groups and GO nanosheets in the membranes matrix. Antifouling ability of membranes was improved after the incorporation of GO nanosheets. In addition, irreversible protein fouling of membranes was substantially decreased with increasing fraction of GO nanosheets (%). The transmission of protein as a function of solution pH and the fraction of GO nanosheets (%) in the membranes was studied for two model proteins (bovine serum albumin; BSA or lysozyme; LYZ). The transmission of BSA or LYZ was controlled by size exclusion and the fraction of GO nanosheets in the membrane matrix. The highest transmission of proteins at their isoelectric points was obtained for membrane containing 2 wt% of GO nanosheets to total weight of polymers.
Introduction
The highly efficient purification and separation of proteins is important in the field of bioseparation engineering, particularly for biotechnology, food, biomedical and pharmaceutical industries.1,2 The complexity of the protein structures, variety of their sequences and folding motives make various approaches inefficient for purification and separation of proteins.3,4 Packed bed column chromatography,5 affinity membrane chromatography,6 adsorption,4 electrophoresis7 and ultrafiltration3,8 have been explored for purification and concentration of proteins from their mixture model solutions and real process samples, with chromatography operations typically being incorporated into downstream processing. Sophisticated chromatography systems are expensive, and a large volume of wastewater containing salts is produced in separation and purification of proteins.5,9 Therefore, an efficient method for purification and separation of proteins with a low operating cost is required. Among the abovementioned separation methods, ultrafiltration (UF) has gained remarkable attention in purification and concentration along with separation of proteins. This method is more efficient, easier to handle and, in distinct contrast to chromatography operations, can be easily manufactured as a single use system and scaled up, at low cost.3,8–10
Polymeric UF membranes are a key component for UF in purification and separation of proteins. UF membranes with an asymmetric porous structure are typically fabricated from polysulfone (PSf), polyethersulfone (PES), poly(arylene ether sulfone) block copolymer, cellulose acetate (CA), polyamide (PA), polyimide (PI) and polyvinylidene fluoride (PVDF) by a non-solvent induced phase separation (NIPS) method.3,11 The major problem in concentration and fractionation of proteins by UF is deterioration in membrane permeability (flux) and selectivity (rejection) due to fouling.12–15 Membrane fouling is affected by many factors, but primary causes have been shown to be the adsorption and deposition of proteins on surface and/or in pores of membranes.3,9,12,16 In more detail, membrane fouling is composed of reversible fouling (Rr) and irreversible fouling (Rir). Reversible protein adsorption or deposition on surface and/or in pores of membranes is responsible for reversible fouling that could be removed by hydraulic cleaning i.e., backwashing and cross flushing. However, irreversible protein adsorption on the surface and/or in pores of membranes leads to irreversible fouling which requires chemical cleaning. Irreversible fouling not only increases the process cost, but also declines membrane lifespan.3,17,18
Significant efforts have been made to overcome this problem and develop mitigation approaches to enhance the performance of UF membranes in ultrafiltration of protein mixture solutions. Four approaches have been exploited to improve antifouling ability and performance of UF membranes in protein mixture solution filtration: (i) post modification of UF membranes by plasma treatment,16 UV induced grafting19 and atom transfer radical polymerization;20 (ii) surface coating of hydrophilic copolymers on the surface of UF membranes;21 (iii) pre-functionalization of hydrophobic polymers via anchoring of hydrophilic functional groups (quaternary ammonium; –CH2N(CH3)3+, –COOH and –SO3H groups) by chemical modification reactions, and their use as additives to fabricate charged blend UF membranes using a NIPS method9,22,23 and (iv) blending of membrane forming polymers with fillers to fabricate low fouling hybrid UF membranes.24 Examples of fillers that have been used successfully in fabrication of low fouling hybrid UF membranes with improved permeability include multiwalled carbon nanotubes,25 sulfonated polyaniline (PANI),12 polymer grafted SiO2 nanoparticles,17 TiO2–ZrO2,18 zeolitic imidazolate framework (ZIF) nanoflakes26 and graphene oxide nanosheets.27 Graphene oxide (GO) nanosheets are considered as single two dimensional carbon sheets in which oxygen-containing functional groups (e.g., hydroxyl, carboxyl, carbonyl, and epoxy groups) make them strongly hydrophilic in nature. GO nanosheets can be used as a filler in the fabrication of low fouling hybrid UF membranes by a NIPS method because of their high hydrophilicity and easy dispersion in polar organic solvents.27–30 Jin et al. have fabricated hybrid UF membranes using PES and GO nanosheets. The water permeability and antifouling ability of hybrid membranes were improved after addition of GO nanosheets. However, the selectivity (rejection capacity) of hybrid membranes at high fraction of GO nanosheets was declined.27
The selectivity of hybrid UF membranes could be improved by anchoring anionic or cationic groups in the barrier layer of membranes.3,8–10,23,31–33 Qiu et al. have fabricated positively charged UF membranes from self-assembly of amphiphilic polystyrene-b-poly-4-vinylpyridine block copolymer. Quaternary ammonium groups in the barrier layer of membranes were anchored by heterogeneous quaternization reaction using 2-chloroacetamide. The selectivity of membranes in separation of bovine serum albumin from haemoglobin in a mixture model solution was enhanced 10 times compared to conventional UF membranes.28 Kumar et al. have reported positively charged organic–inorganic hybrid UF membranes for separation of lysozyme from ovalbumin in a mixture model solution. The best separation of LYZ from OVA in a mixture model solution was achieved at pH = 11 using a high charge density membrane.9 The combination of size and charge based selectivity is ideal for improving the separation performance of membranes in UF of protein mixture solutions. The transport of proteins across the charged UF membranes is also dependent on the type and strength of electrostatic interactions between the membrane and the protein at a specific solution pH.3,9,10,32,33
Thus, the selective separation of proteins using charged hybrid UF membranes is, in principle, possible at a controlled pH and applied transmembrane pressure. To the best of our knowledge, the fabrication of positively charged hybrid UF membranes with improved permeability and antifouling ability based on quaternized polysulfone and GO nanosheets has not been reported previously. In this study, efforts have been made to fabricate hybrid UF membranes with varied charge density from blends of PSf and QPSf polymers with GO nanosheets by solution casting and non-solvent induced phase separation (“phase inversion”) method. The fabricated membranes have been characterized comprehensively and then used in UF of model protein (BSA and LYZ) solution at varied pH and constant applied transmembrane pressure.
Experimental section
Materials
Polysulfone P-3500 was supplied by Solvay Speciality Polymers, Belgium. Expandable graphite flakes with average flake size >500 μm were obtained from Asbury Graphite Mills, USA. Paraformaldehyde ((HCHO)n), chlorotrimethylsilane ((CH3)3SiCl), tin(IV) chloride (SnCl4), potassium permanganate (KMnO4), deuterated chloroform (CDCl3) and hydrogen peroxide (H2O2; 30%) were received from Sigma-Aldrich. Bovine serum albumin was purchased from Sigma-Aldrich and lysozyme was obtained from Fluka Chemicals. Trimethylamine ((CH3)3N), N,N-dimethylacetamide (DMAc), N-methyl-2-pyrrolidone (NMP), diethyl ether, sodium dihydrogen phosphate, disodium hydrogen phosphate and sodium hydroxide (NaOH) were procured from Merck Chemicals. Chloroform (CHCl3), methanol, sulphuric acid (H2SO4) and hydrochloric acid (HCl) were purchased from VWR Chemicals. Other chemicals and reagents were commercial grade and used as received. Distilled water (DW) and DI water (water purified with a Milli-Q system (Millipore)) were used in this study.
Synthesis of quaternized polysulfone
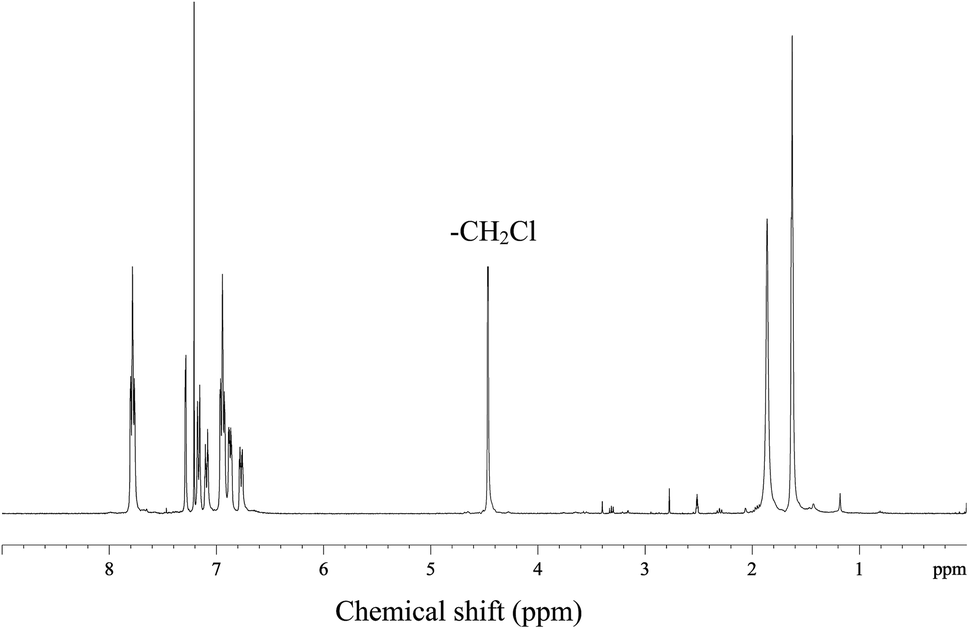
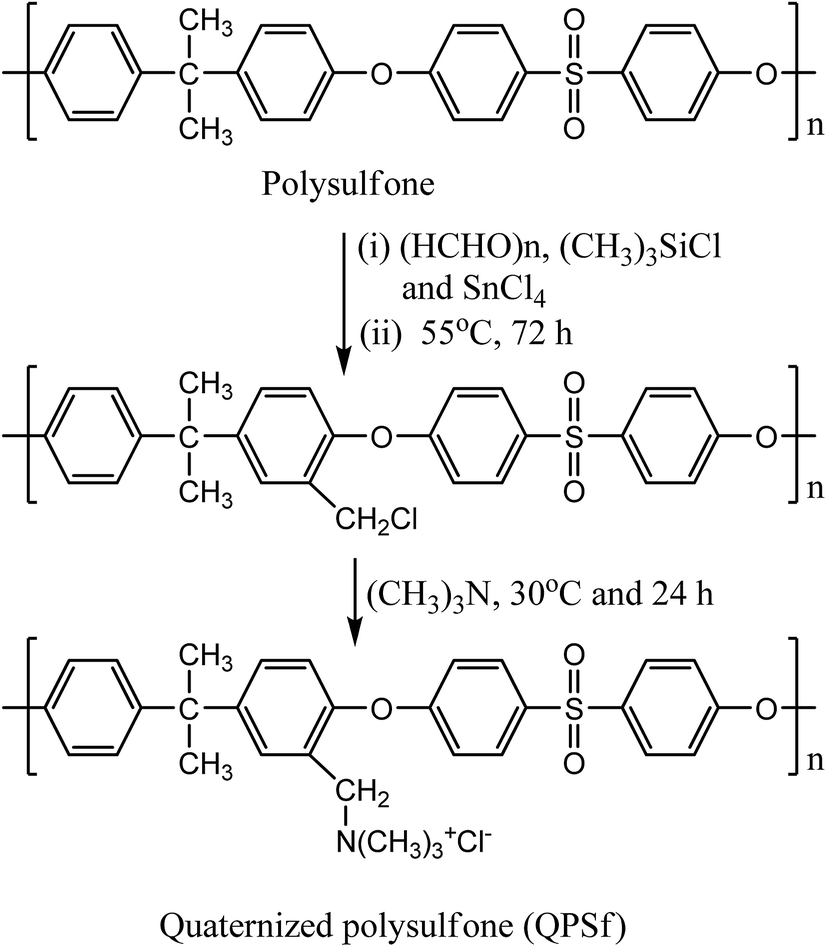
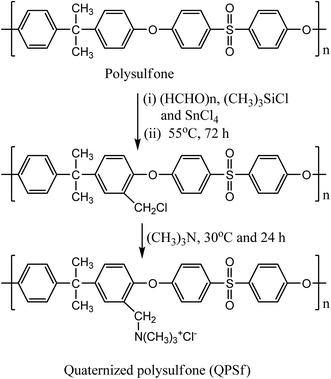
Quaternized polysulfone (QPSf) was synthesized from chloromethylated polysulfone (PSf-CH2Cl) via an in situ quaternization reaction at 40 °C. PSf-CH2Cl was synthesized by chloromethylation reaction using paraformaldehyde, chlorotrimethylsilane and tin(IV) chloride as a catalyst.34 The typical procedure for synthesis of PSf-CH2Cl was as follows: 5 g PSf was dissolved in 250 ml CHCl3 (AR grade, stored in presence of 4 Å molecular sieves) in a round bottom flask equipped with a reflux condenser and magnetic stirrer. After dissolution of PSf, 3.5 g (115 mmol) of paraformaldehyde and 12.5 g (115 mmol) of (CH3)3SiCl were alternatively added. Subsequently, 0.4 ml (1.5 mmol) tin(IV) chloride in 10 ml CHCl3 was slowly added to the reaction mixture solution under stirring condition at 55 °C and the reaction was continued for 72 h at 55 °C. The resulting mixture solution was then precipitated in 500 ml methanol and the precipitated polymer was collected on filter paper by vacuum filtration. The PSf-CH2Cl was again dissolved in 50 ml CHCl3 and then precipitated in 250 ml methanol to remove traces of impurities. A white coloured powder was obtained, which was dried in a vacuum oven at 40 °C for 12 h. The 1H-NMR spectrum for PSf-CH2Cl was recorded in CDCl3 solvent (Fig. 1). The peak at 4.56 ppm corresponds to –CH2Cl group, confirmed successful synthesis of PSf-CH2Cl (cf.Scheme 1). The degree of chloromethylation for PSf was determined according to the previously reported method.35 Details of method is given in Section S1, ESI.† The degree of chloromethylation for PSf was found to be 45.6%.
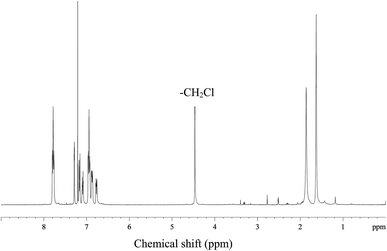 |
| | Fig. 1 The 1H-NMR spectrum for chloromethylated polysulfone (PSf-CH2Cl) in CDCl3. | |
 |
| | Scheme 1 Reaction route for synthesis of quaternized polysulfone via chloromethylation and in situ quaternization reaction using trimethylamine. | |
Quaternized polysulfone was synthesized by in situ quaternization of PSf-CH2Cl with trimethylamine (TMA) (Scheme 1). The typical procedure for synthesis of QPSf was as follows: 1 g PSf-CH2Cl was dissolved in a round bottom flask containing 10 ml DMAc and then 1 ml TMA solution was added drop wise under stirring condition to avoid precipitation. Thereafter, the temperature was raised to 40 °C and the reaction continued for 24 h at constant temperature. The reaction mixture was then poured drop wise into 50 ml diethyl ether and the precipitated polymer in solvent was left over night to remove unreacted TMA. Finally, the purified QPSf was collected on filter paper by vacuum filtration and dried in a vacuum oven at 40 °C for 12 h. FTIR spectrum for QPSf was recorded (Fig. S1, ESI†) to confirm in situ quaternization of PSf-CH2Cl with TMA. The characteristic absorption bands at 3396, 1665 and 1485 cm−1 are observed due to –CH2N(CH3)3+ groups in QPSf and the obtained results confirm in situ quaternization of PSf-CH2Cl with TMA.25,31,34
Synthesis of graphene oxide nanosheets
Graphene oxide nanosheets were synthesized from expandable graphite flakes by a modified Hummers method.27,28,36 The detailed procedure for synthesis of GO nanosheets is given in Section S2, ESI.† FTIR spectrum for GO nanosheets was recorded as depicted in Fig. S2, ESI.† The broad absorption band at 3270 cm−1 is attributed to O–H stretching vibration.27,28 The absorption bands at 1723 and 1615 cm−1 are obtained due to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and aromatic CC stretching vibrations. The absorption bands at 1222 and 1047 cm−1 are ascribed to epoxy C–O and alkoxy C–O stretching vibrations.27,29,30 The results confirm successful synthesis of GO nanosheets by a modified Hummers method.
O and aromatic CC stretching vibrations. The absorption bands at 1222 and 1047 cm−1 are ascribed to epoxy C–O and alkoxy C–O stretching vibrations.27,29,30 The results confirm successful synthesis of GO nanosheets by a modified Hummers method.
Membrane fabrication and characterization
In this study, PSf/QPSf and PSf/QPSf/GO hybrid membranes were fabricated by solution casting and phase inversion method.25,27,29 Predetermined amounts of GO nanosheets were dispersed into NMP by sonication, then, 16 wt% of the dried PSf and QPSf (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) was added into the suspension of GO nanosheets in NMP and stirring continued at 50 °C until polymers dissolved completely. The blend solutions were then sonicated for 30 min and left at room temperature (RT) without stirring to remove any trapped air bubbles. The obtained blend solutions containing PSf/QPSf/GO nanosheets were then cast onto a glass plate using a casting knife with gap height of 250 μm. The proto-membrane film with glass plate was left for 30 s and subsequently, submerged in a DW water coagulation bath until the membrane peeled off from the glass plate. Membranes of ∼125 μm thickness in wet condition were obtained, which were thoroughly washed with DI water to remove traces of NMP. The base membrane was also fabricated in the same way without GO nanosheets. The composition of casting solutions for fabrication of all membranes is included in Table S1, ESI.† The fabricated membranes are designated as membrane AG-X (X being the weight percent (%) of GO nanosheets to total weight of polymers blend i.e. AG-0, AG-1, AG-2 and AG-5).
:1) was added into the suspension of GO nanosheets in NMP and stirring continued at 50 °C until polymers dissolved completely. The blend solutions were then sonicated for 30 min and left at room temperature (RT) without stirring to remove any trapped air bubbles. The obtained blend solutions containing PSf/QPSf/GO nanosheets were then cast onto a glass plate using a casting knife with gap height of 250 μm. The proto-membrane film with glass plate was left for 30 s and subsequently, submerged in a DW water coagulation bath until the membrane peeled off from the glass plate. Membranes of ∼125 μm thickness in wet condition were obtained, which were thoroughly washed with DI water to remove traces of NMP. The base membrane was also fabricated in the same way without GO nanosheets. The composition of casting solutions for fabrication of all membranes is included in Table S1, ESI.† The fabricated membranes are designated as membrane AG-X (X being the weight percent (%) of GO nanosheets to total weight of polymers blend i.e. AG-0, AG-1, AG-2 and AG-5).
The water uptake (φ), porosity (ε), ion-exchange capacity (IEC) and fixed ion concentration (Af) of membranes were determined according to previously reported methods.9,18,37 The details of methods are given in Sections S3 and S4, ESI.† The detailed procedure for determination of free water (φf) and bound water (φb) of membranes is also provided in Section S5, ESI.† The attenuated total reflection-Fourier transform infrared (ATR-FTIR) spectrum for membrane AG-5 was recorded on a Perkin Elmer Spectrum100 spectrometer. The spectrum was recorded over a wide range from 650 to 4000 cm−1 with 32 scans at a resolution of ±4 cm−1. The SurPass Electrokinetic Analyser (Anton Paar GmbH, Austria) was used to measure outer surface zeta potential of membranes and the details of experimental conditions are described in previously reported papers.30,31 The outer surface zeta potential (ζ) of membranes was calculated using Helmholtz–Smoluchowsky equation (eqn (1)):30,31
| | 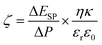 | (1) |
where Δ
ESP/Δ
P is the change in streaming potential with pressure,
η is the electrolyte solution viscosity,
κ is the conductivity of electrolyte solution;
ε0 is the permittivity of free space and
εr is the permittivity of electrolyte solution.
The surface and cross-section morphologies of membranes were observed using a Hitachi S3400N, UK, scanning electron microscope at an acceleration voltage of 20 KV using a secondary electron detector. Membrane samples were flash-frozen using liquid nitrogen and broken to achieve an even cross-section, these were then mounted onto stainless steel stubs and sputter coated with gold (60 s, 40 μA) to create a conductive surface. Water contact angle of membranes was determined on FTÅ 200 contact angle analyzer (First Ten Angstroms, Inc., USA) equipped with video capture by a sessile drop method.9,29,30 To minimize the experimental error, the contact angle was measured at five locations for each membrane and the average value was then reported. In addition, the free energy of interaction at interface between the liquid and the membrane surface (−ΔGSL) was calculated using the Young–Dupre equation (eqn (2)):9,25
| | | −ΔGSL = (1 + cosθ)γTL | (2) |
where
θ is the measured water contact angle and
γTL is the total surface tension of water (72.8 mJ m
−2).
25 The tensile strength and percentage elongation at break point of membranes were determined using a Zwick Z005 displacement controlled tensile testing machine (Zwick-Roell, Germany) at a crosshead speed of 2 mm min
−1 and the details of the operating procedure are given in Section S6, ESI.
†
Pure water flux measurement
A dead-end stirred ultrafiltration cell (Amicon 8200; Millipore Co., USA) connected with a N2 gas cylinder and solution reservoir was used to determine pure water flux of membranes. Each membrane was initially compacted by filtering DI water for 30 min at 2 bar and then the pressure was released to 1 bar. Thereafter, DI water was passed through the membranes for 1 h at 1 bar applied pressure. The mass of collected permeate was measured on a digital balance (Ohaus Adventurer™ Pro Balance, UK). The pure water flux (Jw; L m−2 h−1) was calculated using eqn (3):9,30,38| | 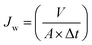 | (3) |
where V is the volume of the collected permeate (L), A is the effective membrane area (m2), and Δt is the permeation time (h).
Antifouling performance
The antifouling ability of membranes with protein solution was evaluated in detail by conducting protein adsorption and filtration experiments at known pH and constant applied transmembrane pressure. In this study, BSA was selected as a model protein to evaluate antifouling ability of membranes. Static protein adsorption experiments were conducted to determine the adsorbed amount of BSA on the membranes at pH = 3 and 7. Circular pieces of membranes (diameter 2.5 cm) were placed into conical flasks containing 25 ml solution of BSA (1 g L−1) in 10 mmol phosphate buffer. Subsequently, the pH of the solutions was adjusted to 3 and 7 using 1 M HCl/NaOH aqueous solutions. The conical flasks were then placed on a shaker at room temperature (RT) for 8 h with a stirring speed of 100 rpm. The concentration of BSA in the supernatant solutions was determined using a Cary 50 Bio UV-Vis spectrophotometer (Varian Inc., USA) at a wavelength of 280 nm. The adsorbed amount of BSA per unit area of membrane (Q; μg cm−2) was calculated using eqn (4):39| | 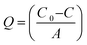 | (4) |
where C0 and C are the initial and final concentration of BSA in solution (μg) and A is the effective membrane area (cm2). Both sides of the membranes were in contact with protein solution; hence the data refer to protein binding at the outer and accessible inner surface of membranes, hence the effective membrane area used was twice the membrane surface area. The reported data are the mean values of triplicate samples for each membrane.
In order to evaluate the contribution of reversible fouling, 500 ml BSA (1 g L−1) solution of pH = 3 was filled into the UF cell. The filtration of BSA solution was performed for 2 h at 1 bar with a stirring speed of 400 rpm. The flux of BSA solution (Jp; L m−2 h−1) was determined from the collected permeate protein solution over 2 h. The membranes were then removed from UF cell and thoroughly washed with DW water. The cleaned membranes were replaced into the cell which was refilled with DI water. The water flux of the cleaned membranes was recorded by passing DI water for 30 min at 1 bar. The flux recovery ratio (FRR, in %) of membranes was determined using eqn (5):9,18,27,29
| | 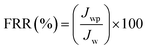 | (5) |
where
Jwp is the water flux of the cleaned membrane after filtration of BSA solution and
Jw is the initial pure water flux of membrane. The following equations were used to evaluate the fouling mechanism in detail. The total fouling ratio (
Rt), reversible fouling ratio (
Rr) and irreversible fouling ratio (
Rir) were calculated using
eqn (6)–(8):
14,25,29| | 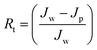 | (6) |
| | 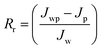 | (7) |
| | 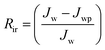 | (8) |
where
Jp is the protein solution flux of membrane and the other terms are as described above.
Ultrafiltration of protein solutions
Ultrafiltration of protein (BSA or LYZ) solution through the membranes was conducted at pH = 5, 7 and 11. 250 ml solution of BSA or LYZ (1 g L−1) of known pH was filled into the reservoir and then ultrafiltration experiments were conducted for 1 h at 1 bar. The concentration of BSA or LYZ in the feed and the permeate solutions was determined at a wavelength of 280 nm using a Cary 50 Bio UV-Vis spectrophotometer. The observed transmission of protein (τobs) through the membranes was calculated using eqn (9):9,25,40| | 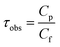 | (9) |
where Cp is the concentration of protein (BSA or LYZ) in the permeate solution after filtration and Cf is the concentration of protein in the feed solution before filtration.
Results and discussion
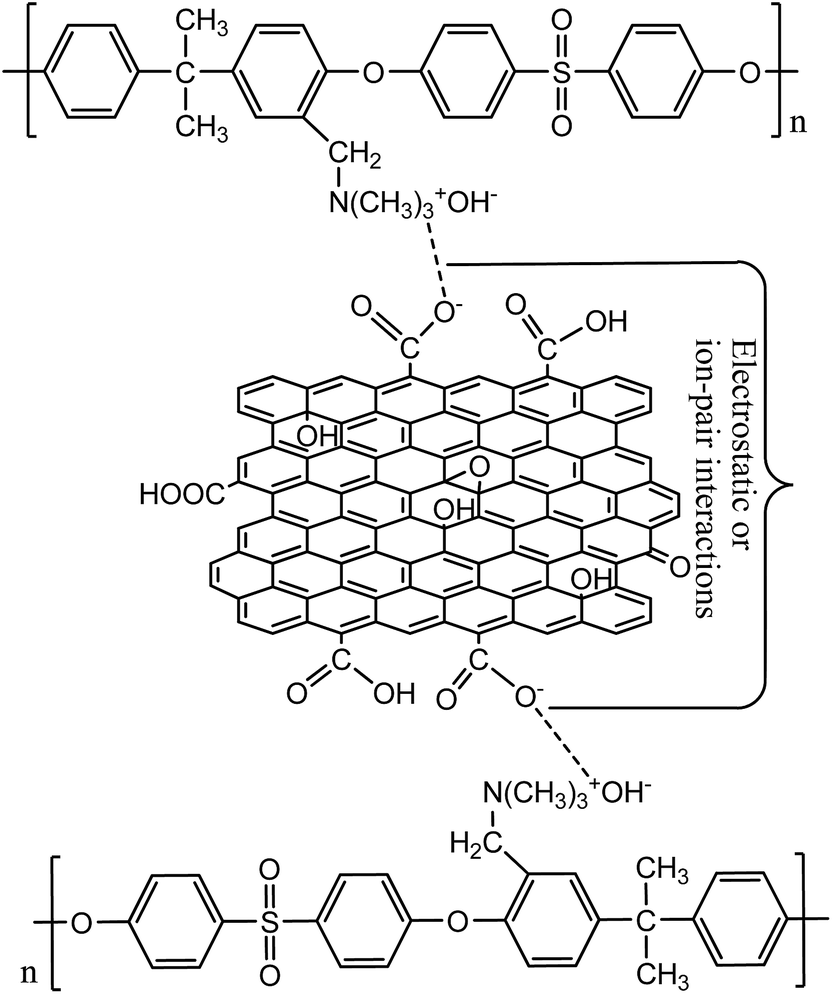
Membranes with varied fraction of GO nanosheets (%) were fabricated from GO and a blend of PSf and QPSf polymers by a NIPS method. GO nanosheets in the membrane matrix were entrapped via electrostatic and/or ion-pair interactions between –CH2N(CH3)3+ groups of QPSf chains and –COO− groups of GO nanosheets (Fig. 2). A maximum of 5 wt% GO nanosheets to total weight of polymer blend (PSf and QPSf) were embedded in the membranes. Efforts were rendered to increase the loading of GO nanosheets from 5 to 7 wt% in the membranes, but pinholes were created after precipitation in DI water coagulation bath and the membranes became mechanically unstable. Due to these reasons, the fraction of GO nanosheets was not increased above 5 wt% in the hybrid membranes.
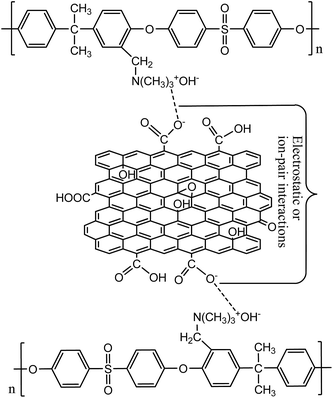 |
| | Fig. 2 Schematic presentation for electrostatic and/-or ion-pair interactions between QPSf and GO nanosheets in the hybrid membranes. | |
Physicochemical and instrumental characterizations
The membranes were characterized physicochemically by determining their water uptake, porosity, ion-exchange capacity and fixed ion concentration (Table 1). It can be seen that φ values increased systematically with fraction of GO nanosheets in the membrane matrix. This could be attributed to (i) the entrapment of water molecules in the membrane matrix via hydrogen bonding interactions with the functional groups (–COOH, OH and –CH2N(CH3)3+) of the membranes25,28,30 and (ii) the formation of tight hydration layer due to spontaneous migration of oxygen-containing groups (–COOH and –OH) of GO nanosheets onto the membrane surface during the phase inversion process.14,27–29 The migration of GO nanosheets onto the membrane surface was also verified from the change in membrane top layer colour with fraction of GO nanosheets (cf. Fig. S3, ESI†). The highest φ value (69.6%) was achieved for membrane AG-5 because of high water capturing ability of this membrane. The data confirm that the hydrophilicity of membranes was enhanced after the incorporation of GO nanosheets.
Table 1 Physicochemical properties of positively charged hybrid membranes: water uptake; φ, free water; φf, bound water; φb, porosity; ε, ion-exchange capacity; IEC and fixed ion concentration; Af
| Membrane |
φ (%) |
φ
f (%) |
φ
b (%) |
ε (%) |
IEC (mequiv. g−1) |
A
f (mequiv. g−1 H2O) |
| AG-0 |
56.8 |
44.6 |
12.2 |
59.3 |
0.68 |
1.19 |
| AG-1 |
62.3 |
46.8 |
15.8 |
68.4 |
0.67 |
1.08 |
| AG-2 |
64.2 |
47.3 |
16.9 |
72.6 |
0.65 |
1.02 |
| AG-5 |
69.6 |
48.1 |
21.5 |
70.2 |
0.60 |
0.86 |
DSC thermograms for the membranes in water swollen state are depicted in Fig. S4, ESI.† One peak was obtained at ∼0 °C for all membranes; this was observed due to the presence of free water in the membranes. The free water content (φf; %) in the membranes was estimated using eqn (10):9,41
| | 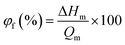 | (10) |
where Δ
Hm is the enthalpy of melting for membrane and its value was obtained from the integration area of the melting enthalpy peak for each membrane in fully swollen state, and
Qm is the melting enthalpy of water at 0 °C (334 J g
−1).
9,41 The bound water content (
φb; %) was calculated from the difference in total water (
φ; %) and free water content (
φf; %) of membranes. The obtained values of
φf and
φb for all membranes are tabulated in
Table 1. The bound water fraction substantially increased from 12.2 to 21.5% for the hybrid membranes with fraction of GO nanosheets up to 5 wt%. This could be attributed to proportional increase in extent of –COOH/–OH groups in the membrane matrix, which are responsible for binding of water molecules in the channels/pores of membranes.
14,27,29 The highest bound water fraction (21.5%) was achieved for membrane AG-5 because more water molecules were entrapped in the channels/pores of membranes. The porosity values for membranes are presented in
Table 1. The porosity of membranes increased with fraction of GO nanosheets up to 2 wt% and the highest
ε (72.6%) value was obtained for membrane AG-2. However, a slight decrease in the
ε value for membrane AG-5 was observed (
cf.Table 1). This can be explained on the basis of hydrophilic effect of GO nanosheets and the viscosity of membrane casting solution during phase inversion process.
14,25,27–30 The porosity of membranes at low fraction of GO nanosheets (up to 2 wt%) in the membrane matrix was high because the rate of solvent exchange with non-solvent during phase-inversion process could be enhanced by hydrophilic GO nanosheets with matrix of polymers (PSf and QPSf) blend.
14,25,27,28 Meanwhile, an excess fraction of GO nanosheets (5 wt%) could be responsible for decline in porosity of membrane AG-5. The ion-exchange capacity and fixed ion concentration values were slightly reduced with fraction of GO nanosheets (wt%) (
Table 1). This is because of proportional decline in extent of –CH
2N(CH
3)
3+ groups in the membrane matrix
via the electrostatic or ion-pair interactions between –CH
2N(CH
3)
3+ groups of QPSf polymer and –COOH
− of GO (
cf.Fig. 2). The IEC of membrane AG-5 was found to be 0.60 mequiv. g
−1, which is lower than that of other membranes in this study (
cf.Table 1). The obtained data for IEC and
Af confirm the mildly charged nature of the membranes. The IEC of all membranes was lower than, for instance, those of other reported positively charged membranes in literature.
35,42–44
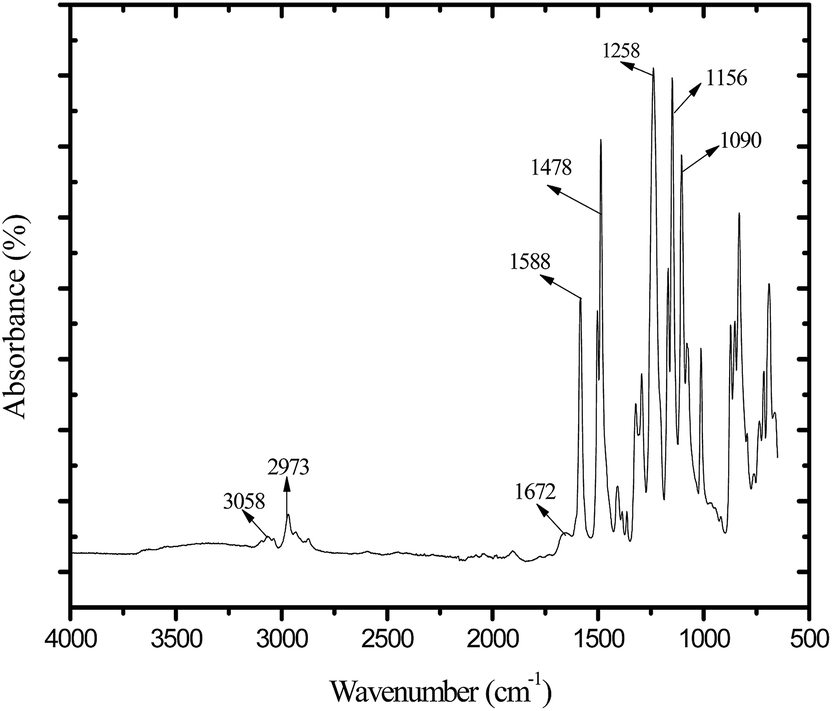
ATR-FTIR spectrum for membrane AG-5 is presented in Fig. 3. The broad peak in the range 3490 to 3330 cm−1 is owing to the stretching vibration of –OH groups and residual water present in the membrane matrix.9,25,45 The absorption bands at 3058 and 2973 cm−1 are obtained due to aromatic and aliphatic stretching vibrations of –CH2 groups.25,27,31,45 The absorption bands at 1258, 1150 and 1082 cm−1 are assigned to the symmetric and asymmetric stretching vibrations of –SO present in the backbone of polymer chains.45 The peaks at 1588 and 1478 cm−1 are assigned to the stretching vibration of aromatic hydrocarbons.25,31,45 The absorption band in the range from 1697 to 1670 cm−1 is attributed to the stretching vibrations of –CO in GO nanosheets.27,30 In addition, the absorption band at 1670 cm−1 is ascribed to presence of –CH2N(CH3)3+ groups in the membrane matrix.25,31,45 The obtained results confirm the incorporation of GO nanosheets in the membranes.
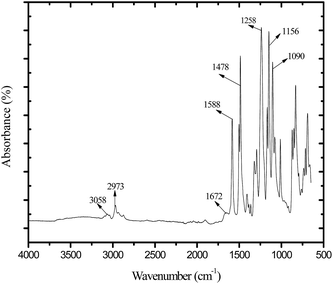 |
| | Fig. 3 ATR-FTIR spectrum for positively charged hybrid membrane AG-5. | |
Tensile tests for the dried membranes were conducted to determine their mechanical strengths. The measured values of stress (MPa) vs. strain (%) are presented in Fig. S5, ESI.† Young's modulus; Emod (MPa), and tensile strength (MPa) values for membranes at the break point were obtained from initial slope of stress versus strain curves (cf. Fig. S5†) and the obtained values are shown in Table 2. Young's modulus and tensile strength (MPa) values for membranes were increased with fraction of GO (%) nanosheets up to 2 wt% and then declined with further addition of GO nanosheets (5 wt%). These results are attributed to (i) high specific area of GO nanosheets, (ii) strong interaction of GO nanosheets with the chains of PSf and QPSf and (iii) aggregation of GO nanosheets in membrane matrix at high fraction.14,27–30 Young's modulus and tensile strength values for membranes were increased up to 2 wt% of GO nanosheets because the adhesion of GO nanosheets at the membrane interface significantly improved due to the high specific area of GO nanosheets, the strong interaction between GO nanosheets and the positively charged polymer chains of QPSf in the membrane matrix. The highest tensile strength (1.84 MPa) and Young's modulus (39.2 MPa) were obtained for membrane AG-2 (cf.Table 2). The excessive fraction of GO nanosheets may have played a role in providing stress convergence points in the membrane, possibly due to the aggregation of GO nanosheets in the membrane AG-5 matrix.14,28,30 Therefore, tensile strength and Young's modulus values for membrane AG-5 were lower than that of all prepared membranes in this study. The tensile strength of the fabricated membranes is higher than, for instance, other reported UF membranes in the literature.15,46,47
Table 2 Tensile properties: Young's modulus; Emod (MPa) and tensile strength (MPa) values for positively charged hybrid membranes with varied fraction of GO nanosheets (%)
| Membrane |
E
mod (MPa) |
Tensile strength (MPa) |
| AG-0 |
28.1 |
1.65 |
| AG-1 |
34.5 |
1.79 |
| AG-2 |
39.2 |
1.84 |
| AG-5 |
27.8 |
1.30 |
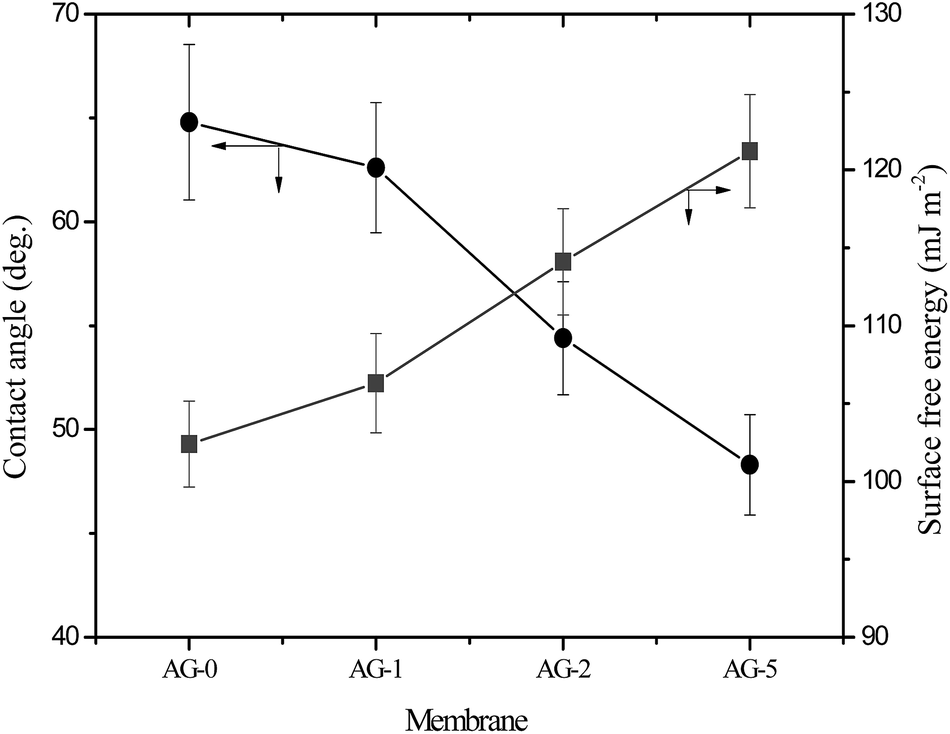
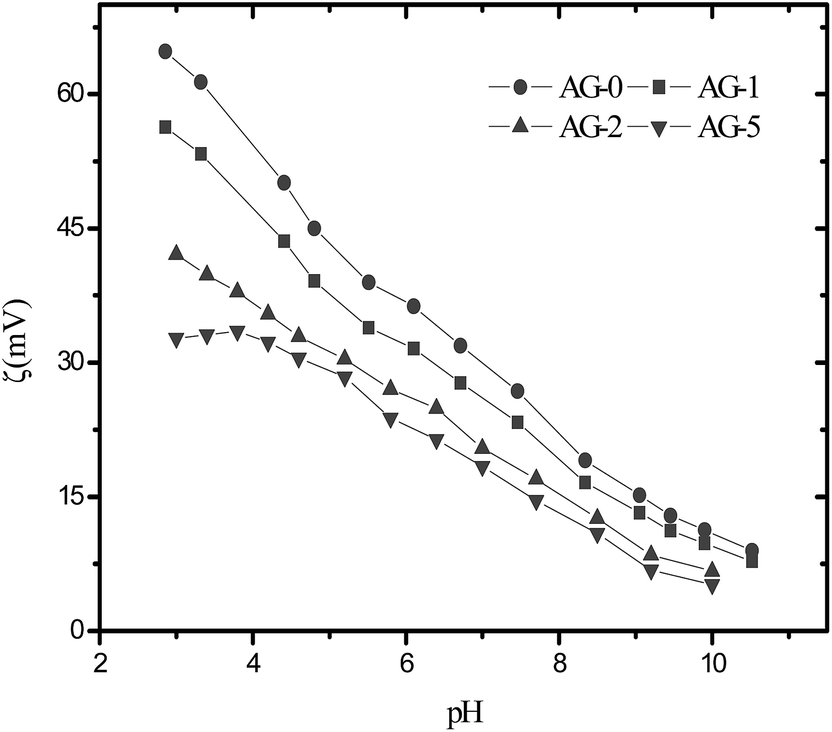
The observed water contact angle and the surface free energy values for membranes are presented in Fig. 4. The water contact angle and the surface free energy values depended on fraction of GO nanosheets. The highest value (65°) of water contact angle and the lowest surface free energy (102.4 mJ m−2) were obtained for membrane AG-0. The water contact angle values decreased and the surface free energies values for membranes increased with fraction of GO nanosheets (cf.Fig. 4). This is owing to the formation of a tight hydration layer on membrane surface via hydrogen bonding interactions between –CH2N(CH3)3+ groups and H2O as well as an additional water binding capacity of –COOH/–OH of GO nanosheets.9,25–28,48 The lowest water contact angle (48°) and highest surface free energy (121 mJ m−2) were obtained for membrane AG-5, correlated with its highest hydrophilicity as assumed from water uptake and bound water fraction values (cf.Table 1). The outer surface zeta potential (ζ) values for membranes at varied fraction of GO nanosheets are presented in Fig. 5. The outer surface ζ values for all membranes were positive over the studied pH range from 3 to 10 because quaternary ammonium groups in the barrier layer of membranes did not allow the deposition of anions in acidic medium.9,25,31,49 The outer surface ζ values for membranes decreased systematically with fraction of GO nanosheets. This is owing to a slight reduction in positive charge density in the barrier layer of membranes (cf.Table 1). Goh et al. and Yu et al., for instance, have reported that the outer surface ζ of positively charged membranes depended on the fraction of –CH2N(CH3)3+ and GO nanosheets in the membranes matrix.48,49
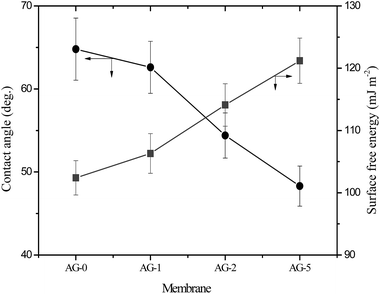 |
| | Fig. 4 Water contact angle and surface free energy for positively charged hybrid membranes with varied fraction of GO nanosheets (%). | |
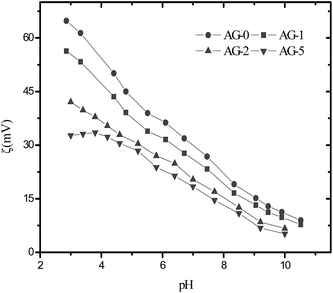 |
| | Fig. 5 The outer surface zeta potential values for positively charged hybrid membranes with varied fraction of GO nanosheets (%). | |
The surface electron microscopy images of membranes are presented in Fig. S6, ESI.† The top surfaces of membranes appear free of microscopic defects. The cross-section electron microscopy images of membranes are shown in Fig. 6. The membranes displayed an asymmetric structure with a dense top (“skin”) layer and a porous supporting layer with fully developed finger-like macrovoids. The cross-section images of membranes were also recorded at low magnifications (cf.Fig. 6) to evaluate if the incorporation of GO nanosheets (%) effected any structural changes in a porous sublayer of membranes. Straight and slightly large finger-like macrovoids were observed when 1 to 2 wt% GO nanosheets to total weight of polymer blend (PSf/QPSf) was added. On the other hand, the finger-like macrovoids in the membrane AG-5 were suppressed with further increase in fraction of GO nanosheets to 5 wt%. Formation of finger-like macrovoids in the sublayer of membranes is promoted by the enhanced hydrophilicity of casting solution at low fraction of carbon fillers (MWCNT-COOH and GO nanosheets).14,25,28,48 Obviously, this phenomenon took place in the formation of membrane AG-1 and AG-2 from casting solutions containing GO nanosheets up to 2 wt%. Furthermore, the viscosity of casting solutions played a major role in the fabrication of asymmetric UF membranes at high fraction of carbon fillers i.e., the porosity and size of finger-like macrovoids are suppressed at high viscosity of casting solutions due to delay in the exchange rate of solvent with non-solvent during the phase inversion process.25,27–29,48 Due to this reason, the porosity (cf.Table 1) and size of the finger-like macrovoids in membrane AG-5 (cf.Fig. 6) were reduced at 5 wt% loading of GO nanosheets. The same trend was observed in the pores of fully developed finger-like macrovoids of membranes (cf. below images in Fig. 6).
 |
| | Fig. 6 Cross-section SEM images at high and low resolution for positively charged hybrid membranes prepared with varied fraction of GO nanosheets (cf. Table S1†): (A) AG-0; (B) AG-1; (C) AG-2 and (D) AG-5. | |
Membrane permeability
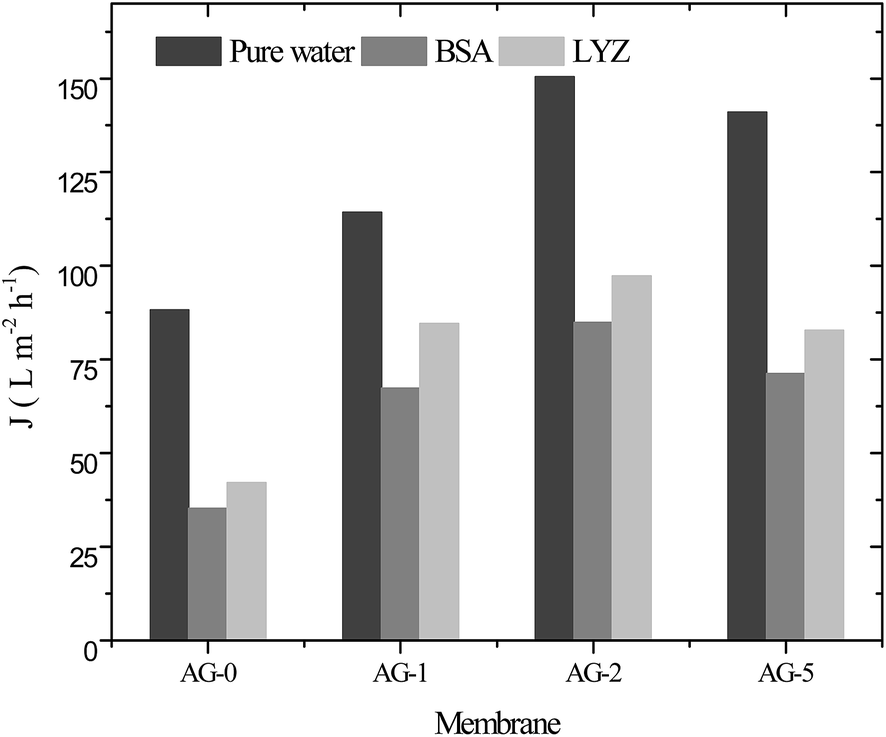
The water flux of membranes was measured to see the effect of GO nanosheets fraction (%) on membrane permeability and the obtained data for all membranes is depicted in Fig. 7. The water flux values increased proportionally to the fraction of GO nanosheets up to 2 wt% and then decreased at fraction of 5 wt% GO nanosheets. These results were associated with increase in hydrophilicity and porosity of the membranes. High hydrophilicity enhanced the membrane permeability by facilitating transport of water molecules through the hybrid membranes. In addition, water is easily passed through highly porous hydrophilic membranes at constant applied transmembrane pressure.9,14,25,30 The water flux (Jw) of membrane AG-0 was found to be 88 L m−2 h−1, which is lower than that of Jw for all other membranes because of its low bound water capacity, hydrophilicity and porosity (cf.Table 1 and Fig. 4). The highest Jw (150 L m−2 h−1) was obtained for membrane AG-2 due to fast transport of water molecules via more hydrophilic and porous membrane as well as wider macrovoids at the bottom (cf.Table 1, Fig. 4 and 6). However, the water flux of membrane AG-5 (140 L m−2 h−1) was lower than that of membrane AG-2. This is correlated to the observed suppression in finger-like macrovoids of membrane sublayer and lower porosity at 5 wt% GO nanosheets loading (cf.Table 1 and Fig. 6). The protein (BSA or LYZ) solution flux (Jp) depended on the fraction of GO nanosheets in the membranes matrix (Fig. 7). As expected based on the water permeability data, Jp values increased substantially with fraction of GO nanosheets up to 2 wt% and then decreased with further increase in fraction of GO nanosheets to 5 wt%. The highest Jp (BSA: 84 L m−2 h−1 and LYZ: 97 L m−2 h−1) values were obtained for membrane AG-2.
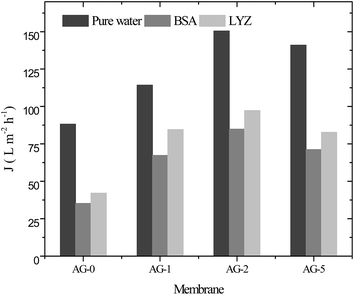 |
| | Fig. 7 Pure water flux and protein (BSA and LYZ) solution flux for positively charged hybrid membranes at varied fraction of GO nanosheets (%), 1 bar applied pressure, pH = 7 and a stirring speed of 400 rpm. | |
It is reported that concentration polarization near the membrane surface during ultrafiltration of protein solution at constant applied transmembrane pressure could be minimized by rigorous stirring.50 In this study, all filtration experiments were performed at a stirring speed of 400 rpm and 1 bar applied transmembrane pressure in an effort to minimize the effect of concentration polarization on decline in protein solution flux with time. Therefore, membrane fouling by protein was mainly responsible for lower Jp values than Jw values for all membranes. The arguments with respect to membrane permeability as function of pore structure should be taken with care because permeability is governed by the structure of top barrier layer which is not easily accessible for direct characterization.9,14,25,36
Antifouling performance
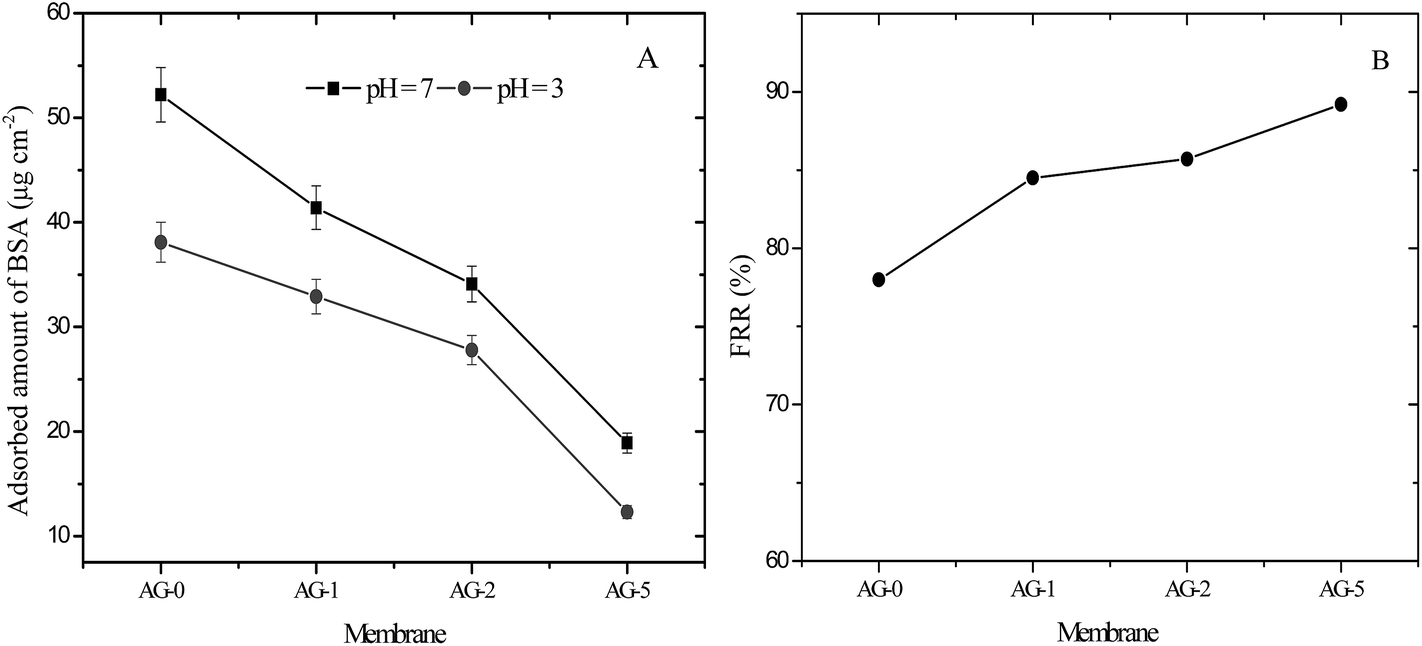
The antifouling performance of membranes was evaluated in terms of protein adsorption and the flux recovery ratio; FRR, total fouling ratio; Rt, reversible fouling ratio; Rr and irreversible ratio; Rir, respectively. In this study, bovine serum albumin (BSA; 67 kg mol−1, isoelectric point; pI = 5) was selected as a model protein to assess the antifouling ability of membranes. The adsorbed amount of BSA on the membranes at pH = 3 and 7 is presented in Fig. 8(A). The adsorbed amounts of BSA at both pH values were decreased significantly for membranes with increasing fraction of GO nanosheets (%). The obtained data is consistent with the observed contact angle and outer surface ζ for membranes at varied fraction of GO nanosheets (cf.Fig. 4 and 5). The effective reduction in the adsorbed protein amount could be due to prevention of BSA molecules accumulation on the membrane surfaces by a tight hydration layer.14,25,27,30 Membrane AG-5 was less prone to BSA adsorption in comparison to all other membranes. The adsorbed BSA amount at pH = 7 for membrane AG-5 was found to be 19 μg cm−2, which is much lower than that for membrane AG-0 (52 μg cm−2). Furthermore, the adsorbed amount of BSA on the membranes was low at pH = 3. The effect of solution pH on decline in adsorption of protein is due to changes in the membrane morphology, hydrophilicity and electrostatic repulsion or interaction between protein and the membrane.10,25,31–33 The net charge on BSA became positive when its solution pH was changed from 7 to 3 and the membrane surface remained positively charged at pH = 3 (cf.Fig. 5). Due to this reason, electrostatic repulsions between positively charged BSA (BSA+) and the membranes occurred at low protein solution pH (pH = 3) and thus protein was excluded by positively charged membranes. However, more protein is excluded by positively charged hybrid membrane with higher hydrophilicity because the presence of a tight hydration layer on the membrane surface does not allow direct contact of protein molecules with membranes.14,25,27,30 This phenomenon was obtained for membranes containing increased amount of GO nanosheets (%). Accordingly, the difference in the adsorbed amounts of BSA among the membranes at pH = 3 may be attributed to the coupling effect of membrane hydrophilicity, membrane charge and membrane morphology. The adsorbed amount of BSA on the fabricated membranes was lower than, for instance, other reported membranes in literature.51–54
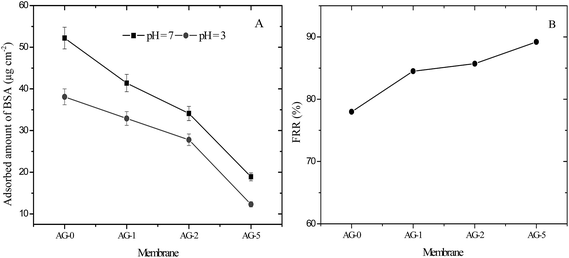 |
| | Fig. 8 (A) Adsorbed amount of BSA on positively charged hybrid membranes at pH = 3 and 7 and (B) FRR values for membranes after ultrafiltration of 500 ml BSA (1 g L−1) solution at pH = 3. | |
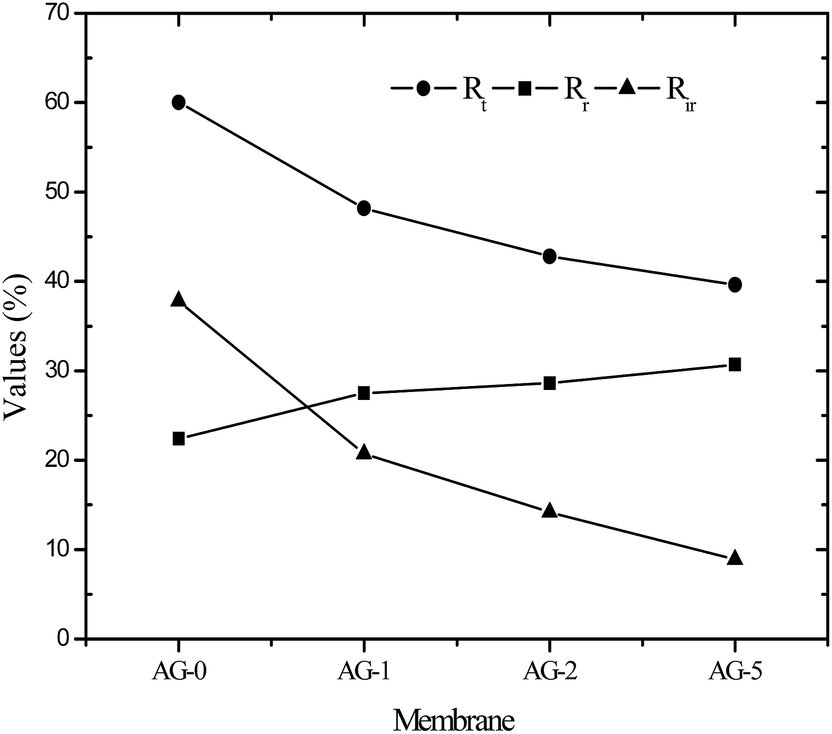
Flux recovery ratio (FRR) values for membranes after UF of BSA solution at pH = 3 are depicted in Fig. 8(B). FRR value for membrane AG-0 (i.e., without GO nanosheets) was lower than that of membranes with GO nanosheets embedded. FRR value for membrane AG-0 was found to be 78%, indicating the membrane fouling was caused by adsorption of protein on the surface and/or in pores via hydrophobic interactions between BSA and unfunctionalized polymer in the membrane matrix.9,25,30,49,52 The membranes containing GO nanosheets exhibited an improvement in FRR values from 78% to 89.2% under similar experimental conditions. This is attributed to the change in membrane hydrophilicity and surface charge density after incorporation of GO nanosheets. The hydration layer on surfaces could prevent direct contact of protein molecules with membranes during filtration of protein solutions.14,27,28,30 The highest FRR value (89.2%) was obtained for membrane AG-5. To understand the fouling behaviour of membranes in detail, total fouling ratio (Rt), reversible fouling ratio (Rr) and irreversible fouling (Rir) ratios were calculated from water flux of membrane before and after UF of BSA solution as well as subsequent cleaning with DI water. The obtained values for membranes are presented in Fig. 9. Rt values for GO nanosheets embedded membranes (AG-1, AG-2, AG-5) were lower than that of membrane AG-0 (i.e., without GO nanosheets). Low Rt values indicated minimal loss in total flux and less fouling in membranes by protein after UF of BSA solution at pH = 3. The Rr values also increased with fraction of GO nanosheets, whereas Rir values decreased from 20.7% to 8.9% with increasing fraction of GO nanosheets (1 wt% to 5 wt%). The Rir values were lower than that of membrane AG-0 for which it was found to be 37.8%. It is therefore clear that incorporation of GO nanosheets decreased irreversible fouling of the hybrid membranes by resisting protein adsorption. Overall, the lowest total fouling and irreversible fouling ratio for membrane AG-5 suggest that the loosely bound protein on surface and/or in pores of membranes could be removed easily by simple cleaning with DI water.
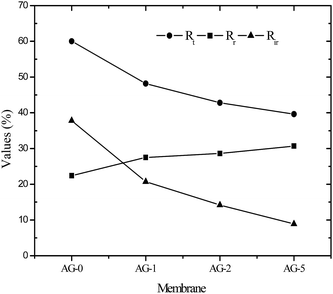 |
| | Fig. 9 Summary of reversible fouling ratio; Rr, irreversible fouling ratio; Rir and total fouling ratio; Rt for positively charged hybrid membranes. | |
Tunable protein ultrafiltration
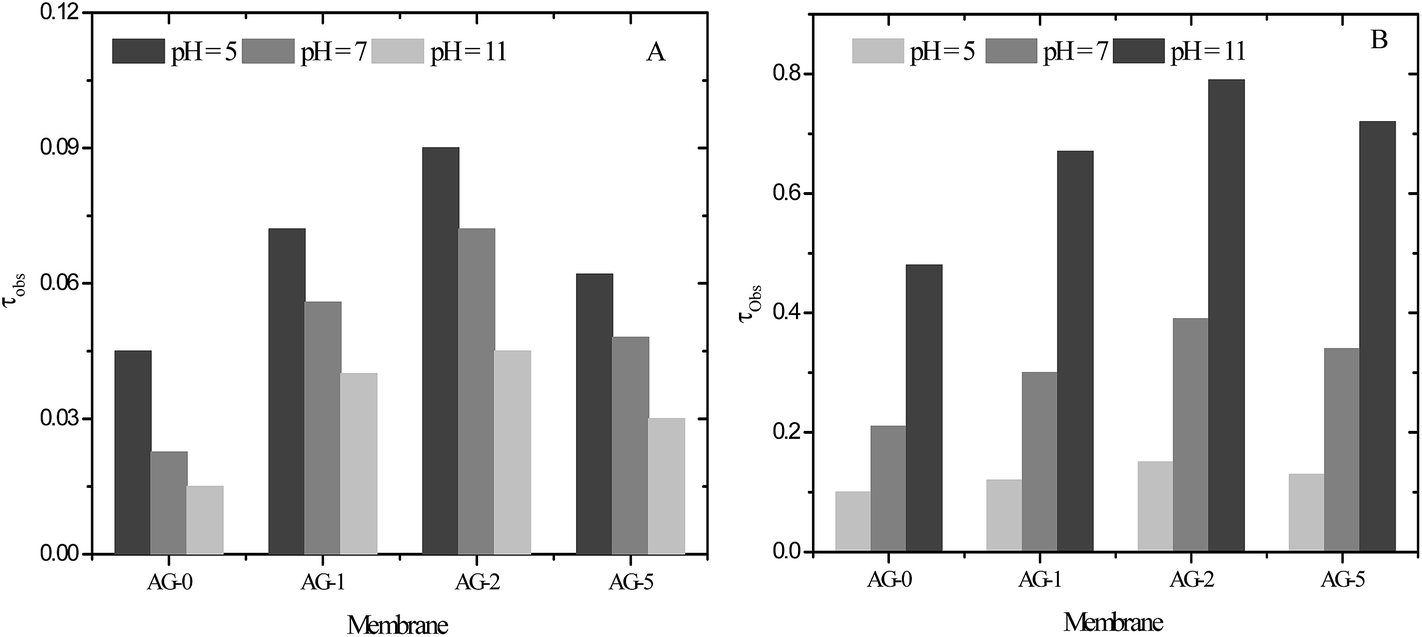
The pH of protein solution plays an important role in UF of protein solutions using charged UF membranes. The electrical charge on the proteins and membranes could be tuned by changing solution pH because of acidic or basic groups of proteins and membranes. The acidic or basic groups ionize as solution pH is changed and attractive or repulsive interactions occur between the protein and the membranes.10,21,25,32,33 In this study, the change in protein charge was considered due to the presence of –CH2N(CH3)3+ groups and GO nanosheets in the barrier layer of membranes. In addition to BSA, lysozyme (LYZ) was selected as a second model protein. The transmission of protein (BSA or LYZ) through the membranes in UF at varied pH and constant applied transmembrane pressure was determined. The obtained τobs values for BSA and LYZ are presented in Fig. 10(A and B).
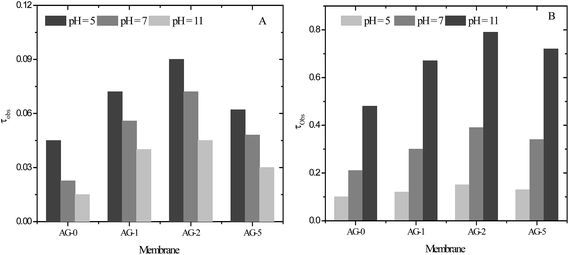 |
| | Fig. 10 The observed transmission (τobs) for: (A) BSA and (B) LYZ (1 g L−1) through positively charged hybrid membranes at varied pH, 1 bar applied transmembrane pressure and a stirring speed of 400 rpm. | |
Transmission of BSA and LYZ across the membrane AG-0 was lower than that of the GO nanosheets embedded membranes AG-1, AG-2 and AG-5. Low τobs values for membrane AG-0 were due to the adsorption or deposition of protein on the less hydrophilic membrane via hydrophobic interactions.9,26,27,30 Thus, the flux of BSA and LYZ across membrane AG-0 were reduced because of pore blocking and a shift in pore size of the effective barrier layer to a lower value.8,19,23,25 The transmission of BSA through the membranes was lower compared to that of LYZ under same experimental conditions (Fig. 10(B)). This is ascribed to the size difference between BSA and LYZ. The size of BSA is larger than LYZ as indicated by their molecular weights and hydrodynamic radii.55,56 Transmission of BSA and LYZ also depended on the fraction of GO nanosheets (%) and protein solution pH. The transmission of BSA and LYZ through the membranes was systematically increased with fraction of GO nanosheets up to 2 wt% and then decreased with further increase in fraction of GO nanosheets to 5 wt%, indicated by the relative τobs values for BSA and LYZ. The highest transmission of BSA and LYZ through all membranes was achieved at pH = 5 and 11, corresponding to pI values of BSA and LYZ, respectively. These results are attributed to the change in porosity and hydrophilicity of membranes with the fraction of GO nanosheets (cf.Table 1 and Fig. 4). The charged UF membranes did not create additional hindrance for the transmission of proteins at their pI values and protein fouling was effectively reduced by the increase in the hydrophilicity of the surface (cf. above). The highest transmission of proteins through the charged UF membranes at their pI values had also been reported by other research groups.8–10,25,32,33,57 The observed transmission of BSA through the membranes was gradually reduced when BSA solution pH changed to 7 and 11. This is due to the formation of a negatively charged self rejecting layer of BSA molecules on the membrane surface via electrostatic interactions between the negatively charged BSA (BSA−) and the positively charged membrane surfaces (cf.Fig. 5). Therefore, the long range electrostatic repulsions between the negatively charged self-rejecting layer of BSA and negatively charged BSA molecules in solution could be responsible for further decline in transmission of BSA at pH > pI of BSA.10,25,30,31,57 At pH = 11, the transmission of BSA through the membrane AG-5 was found to be 0.03, which is higher than that of membrane AG-0 because of its lower charge density and protein retention ability. The transmission of LYZ through the membranes was higher than for BSA at all studied pH values due to its smaller size.56 The τobs values of LYZ for membranes was low at pH = 5 and 7. This was obtained due to the exclusion of positively charged LYZ (LYZ+) by similar charged membranes.8,25,31,33 The transmission of LYZ through the membranes was also influenced by the charge density and fraction of GO nanosheets (%). Transmission of LYZ for all membranes was maximum at pH = 11. The observed transmission of LYZ for membrane AG-2 was found to be 0.79, which is highest among all membranes. In this case, the porosity of membrane AG-2 was more dominant than it's hydrophilicity i.e., LYZ could be easily passed through the highly porous and hydrophilic membrane AG-2 instead of less porous and highly hydrophilic membrane AG-5. Overall, the effect of electrostatic repulsion on the low transmission (high rejection) was much stronger when the fixed charge groups on the membranes were dominant (low pH value). The observed transmission of LYZ for membrane AG-2 was ∼18 times higher than for the transmission of BSA at pH = 11 and 1 bar. These results are suggesting the possible separation of BSA and LYZ from their binary mixture as a model solution at pH = 11 by UF using positively charged hybrid UF membranes.
Conclusions
Low fouling novel positively charged hybrid ultrafiltration membranes were successfully fabricated from blends of PSf and QPSf as well as GO nanosheets by a non-solvent induced phase separation method. The membranes exhibited an anisotropic structure with a relatively dense top (“skin”) layer, a porous sublayer and fully developed finger-like macrovoids. The membrane porosity, hydrophilicity and charge density could be adjusted by varying the fraction of GO nanosheets in the casting solutions at very low values. The hybrid membranes were positively charged over the studied pH range from 3 to 10. Incorporation of GO nanosheets in the membrane matrix containing –CH2N(CH3)3+ groups increased the hydration capacity of membrane surface but also reduced the net charge density. The membranes were less prone to protein adsorption and antifouling ability was further improved after addition of GO nanosheets. The irreversible protein fouling of membranes was substantially decreased with fraction of GO nanosheets. The lowest value of irreversible protein fouling was achieved for membrane AG-5 (i.e., 5 wt% of GO nanosheets to total weight of polymers). Thus, protein adsorption and antifouling ability of membranes could be tuned by varying fraction of GO nanosheets in the casting solutions. The pure water and protein solution fluxes of membranes were controlled by solution pH, fractions of –CH2N(CH3)3+ groups and GO nanosheets in the membrane matrix. The highest pure water and protein fluxes were obtained for the membrane with 2 wt% of GO nanosheets (membrane AG-2). This maximum as function of varied fraction of GO nanosheets correlated with that for volume porosity and trends in top layer and cross-section morphology observed with electron microscopy, although the true maximum may lie at a fraction not directly examined in this study. The highest transmission of BSA and LYZ through the membranes were achieved at pH = 5 and 11. These results confirm the suitability of low fouling novel positively charged hybrid membranes in selective separation of proteins by UF isoelectric focusing technology. Moreover, the trade-off between permeability and selectivity which is commonly observed for conventional UF membranes could be overcome efficiently by tuning the fraction of QPSf and GO nanosheets in the membrane matrix to exploit combined size exclusion and charge effects at low fouling.
Acknowledgements
Mahendra Kumar acknowledges the Irish Research Council, Ireland for awarding IRC Postdoctoral Fellowship. Mr Declan McGlade acknowledges the support of the Environmental Protection Agency (EPA STRIVE 2011-W-MS-8). The authors are thankful to Dr Andreas Heise, School of Chemical Sciences, Dublin City University, Dublin, for giving permission to conduct DSC analysis and National Centre for Sensor Research (NCSR) at DCU for access to analytical equipment.
References
-
A. K. Pabby, S. S. H. Rizvi and A. M. Sastre, Handbook of membrane separations: chemical, pharmaceutical, food, and biotechnological applications, CRC Press, 2009 Search PubMed.
- J. Li, X. Liao, Q. Zhang and B. Shi, J. Chromatogr. B, 2013, 928, 131–138 CrossRef CAS PubMed.
- M. Kumar and M. Ulbricht, Polymer, 2014, 55, 354–365 CrossRef CAS.
- S. Yuan, Q. Deng, G. Fang, M. Pan, X. Zhai and S. Wang, J. Mater. Chem., 2012, 22, 3965–3972 RSC.
- T. M. Przybycien, N. S. Pujar and L. M. Steele, Curr. Opin. Biotechnol., 2004, 15, 469–478 CrossRef CAS PubMed.
- S. R. Dods, O. Hardick, B. Stevens and D. G. Bracewel, J. Chromatogr. A, 2015, 1376, 74–83 CrossRef CAS PubMed.
- V. Orr, L. Zhong, M. M. Young and C. P. Cho, Biotechnol. Adv., 2013, 31, 450–465 CrossRef CAS PubMed.
- V. Valino, M. F. S. Roman, R. Ibanez and I. Ortiz, Sep. Purif. Technol., 2014, 125, 163–169 CrossRef CAS.
- M. Kumar, D. McGlade and J. Lawler, RSC Adv., 2014, 4, 21699–21711 RSC.
- A. Arunkumar and M. R. Etzel, J. Membr. Sci., 2014, 454, 488–495 CrossRef CAS.
- M. Ulbricht, Polymer, 2006, 47, 2217–2262 CrossRef CAS.
- B. T. McVerry, J. A. T. Temple, X. Huang, K. L. Marsh, E. M. V. Hoek and R. B. Kaner, Chem. Mater., 2013, 25, 3597–3602 CrossRef CAS.
- Y. Liu, C. Ma, S. Wang, H. Guo, B. Zhang, L. Zhang, K. Gu and J. Gu, RSC Adv., 2015, 5, 21316–21325 RSC.
- S. Zinadini, A. A. Zinatizadeh, M. Rahimi, V. Vatanpour and H. Zangeneh, J. Membr. Sci., 2014, 453, 292–301 CrossRef CAS.
- X. Huang, J. Zhang, W. Wang, Y. Liu, Z. Zhang, L. Li and W. Fan, RSC Adv., 2015, 5, 18258–18266 RSC.
- J. Imbrogno, M. D. Williams and G. Belfort, ACS Appl. Mater. Interfaces, 2015, 7, 2385–2392 CAS.
- L. J. Zhu, L. P. Zhu, Y. F. Zhao, B. K. Zhu and Y. Y. Xu, J. Mater. Chem. A, 2014, 2, 15566–15574 CAS.
- A. Sotto, J. Kim, J. M. Arsuaga, G. del Rosario, A. Martınez, D. Nam, P. Luise and B. van der Bruggen, J. Mater. Chem. A, 2014, 2, 7054–7064 CAS.
- P. D. Peeva, T. Knoche, T. Pieper and M. Ulbricht, Ind. Eng. Chem. Res., 2012, 51, 7231–7241 CrossRef CAS.
- D. Keskin, J. I. Clodt, J. Hahn, V. Abetz and V. Filiz, Langmuir, 2014, 30, 8907–8914 CrossRef CAS PubMed.
- N. J. Lin, H. S. Yang, Y. Chang, K. L. Tung, W. H. Chen, H. W. Cheng, S. W. Hsiao, P. Aimar, K. Yamamoto and J. Y. Lai, Langmuir, 2013, 29, 10183–10193 CrossRef CAS PubMed.
- Y. H. Cho, J. Han, S. Han, M. D. Guiver and H. B. Park, J. Membr. Sci., 2013, 445, 220–227 CrossRef CAS.
- P. Kanagaraj, S. Neelakandan, A. Nagendran, D. Rana, T. Matsuura and A. Muthumeenal, RSC Adv., 2015, 5, 27594–27602 RSC.
- P. S. Goh, B. C. Ng, W. J. Lau and A. F. Ismail, Sep. Purif. Rev., 2015, 44, 216–249 CrossRef CAS.
- M. Kumar and M. Ulbricht, J. Membr. Sci., 2013, 448, 62–73 CrossRef CAS.
- Z. X. Low, A. Razmjou, K. Wang, S. Gray, M. Duke and H. Wang, J. Membr. Sci., 2014, 460, 9–17 CrossRef CAS.
- F. Jin, W. Lv, C. Zhang, Z. Li, R. Su, W. Qi, Q. H. Yang and Z. He, RSC Adv., 2013, 3, 21394–21397 RSC.
- J. Lee, H. R. Chae, Y. J. Won, K. Lee, C. H. Lee, H. H. Lee, I. C. Kim and J. M. Lee, J. Membr. Sci., 2013, 448, 223–230 CrossRef CAS.
- L. Yang, B. Tang and P. Wu, J. Mater. Chem. A, 2014, 2, 18562–18573 CAS.
- H. Zhao, L. Wu, Z. Zhou, L. Zhang and H. Chen, Phys. Chem. Chem. Phys., 2013, 15, 9084–9092 RSC.
- M. Kumar and M. Ulbricht, RSC Adv., 2013, 3, 12190–12203 RSC.
- X. Qiu, H. Yu, M. Karunakaran, N. Pradeep, S. P. Nunes and K. V. Peinemann, ACS Nano, 2013, 7, 768–776 CrossRef CAS PubMed.
- M. Rohani, A. Mehta and A. L. Zydney, J. Membr. Sci., 2010, 362, 434–443 CrossRef CAS.
- P. T. Nonjola, M. K. Mathe and R. M. Modibedi, Int. J. Hydrogen Energy, 2013, 38, 5115–5121 CrossRef CAS.
- F. Zhang, H. Zhang and C. Qu, J. Mater. Chem., 2011, 21, 12744–12752 RSC.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- Y. Zhang, L. Zou, B. P. Ladewig and D. Mulcahy, Desalination, 2015, 362, 59–67 CrossRef CAS.
- E. Eren, A. Sarihan, B. Eren, H. Gumus and F. O. Kocak, J. Membr. Sci., 2015, 475, 1–8 CrossRef CAS.
- C. Xu, S. Gu, J. Huang, W. Xu, H. Xia, J. Du, H. Liu, H. Yang, Y. Zhou and Z. Bai, J. Appl. Polym. Sci., 2015, 132, 41327–41333 Search PubMed.
- R. Ghosh and Z. F. Cui, J. Membr. Sci., 1998, 139, 17–28 CrossRef CAS.
- Z. H. Ping, Q. T. Nguyen, S. M. Chen, J. Q. Zhou and Y. D. Ding, Polymer, 2001, 42, 8461–8467 CrossRef CAS.
- L. Yu, Y. Zhang, Y. Wang, H. Zhang and J. Liu, J. Hazard. Mater., 2015, 287, 373–383 CrossRef CAS PubMed.
- C. G. Morandi, R. Peach, H. M. Krieg and J. Kerres, J. Mater. Chem. A, 2015, 3, 1110–1120 CAS.
- E. Guler, Y. Zhang, M. Saakes and K. Nijmeijer, ChemSusChem, 2012, 5, 2262–2270 CrossRef CAS PubMed.
-
G. Socrates, Infrared characteristic group frequencies, Wiley, New York, 1980 Search PubMed.
- S. Liang, K. Xiao, Y. Mo and X. Huang, J. Membr. Sci., 2012, 394–395, 184–192 CrossRef CAS.
- M. H. Razzaghi, A. Safekordi, M. Tavakolmoghadam, F. Rekabdar and M. Hemmati, J. Membr. Sci., 2014, 470, 547–557 CrossRef.
- K. Goh, L. Setiawan, L. Wei, R. Si, A. G. Fane, R. Wang and Y. Che, J. Membr. Sci., 2015, 474, 244–253 CrossRef CAS.
- L. Yu, Y. Zhang, H. Zhang and J. Liu, Desalination, 2015, 359, 176–185 CrossRef CAS.
- T. Wang, Y. Q. Wang, Y. L. Su and Z. Y. Jiang, J. Membr. Sci., 2006, 280, 343–350 CrossRef CAS.
- M. K. Sinha and M. K. Purkait, RSC Adv., 2015, 5, 22609–22619 RSC.
- A. Venault, Y. Chang, H. S. Yang, P. Y. Lin, Y. J. Shih and A. Higuchi, J. Membr. Sci., 2014, 454, 253–263 CrossRef CAS.
- E. Celik, L. Liu and H. Choi, Water Res., 2011, 45, 5287–5294 CrossRef CAS PubMed.
- C. Zhao, X. Xu, J. Chen and F. Yang, J. Environ. Chem. Eng., 2013, 1, 349–354 CrossRef CAS.
- Z. Wang, X. Yao and Y. Wang, J. Mater. Chem., 2012, 22, 20542–20548 RSC.
- H. Y. Huang, T. J. Lo, Y. C. Chen, J. B. Hu and P. L. Urban, Anal. Methods, 2013, 5, 5908–5911 RSC.
- M. Sorci, M. Gu, C. L. Heldt, E. Grafeld and G. Belfort, Biotechnol. Bioeng., 2013, 110, 1704–1713 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06893b |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a