
Effects of non covalent interactions in light emitting properties of bis-pyridyl-alkyl-di-imines†
Fayaz Baiga,
Rajni Kantb,
Vivek K. Guptab and
Madhushree Sarkar*a
aDepartment of Chemistry, Birla Institute of Technology and Science, Pilani Campus, Pilani, Rajasthan, India. E-mail: msarkar@pilani.bits-pilani.ac.in; Fax: +91-1596-244183; Tel: +91-1596-515679
bPost Graduate Department of Physics, University of Jammu, Jammu Tawi, India. E-mail: rkvk.paper11@gmail.com; Fax: +91-191-2432051; Tel: +91-191-2432051
First published on 2nd June 2015
Abstract
The crystal structures of three bis-pyridyl-alkyl-di-imines are studied. The non-covalent interactions lead to the packing of the crystals of the compounds in such a way that crystallization induced emission (CIE) is observed for bis-pyridyl-ethyl-di-imine and bis-pyridyl-butyl-di-imine, while for bis-pyridyl-di-imines, with no alkyl spacer, the luminescence is absent in the solid state. The enhanced emission in the solid state is attributed to the collective effects of planarization and the specific arrangement of molecules.
Introduction
The light-emitting molecular solids and their potential utility in optical devices such as organic light-emitting diodes (OLEDs),1 sensors,2a–d photovoltaic cells2e and biological imaging2f,2g has emerged as one of the most promising areas of research in the past few years. The optical devices should be able to function in the solid state. Therefore, in principle an ideal luminescent material must possess high luminescence efficiency in the solid state. There has been an enormous increase in research studies on the luminescence in the aggregated state. In 2001, Tang and coworkers first noticed the aggregation based emission (AIE) and explored new kinds of luminescent materials.3 The AIE property in luminescent materials involves the enhancement in fluorescence intensities, when the molecules form aggregates/clusters. Tang and coworkers reported a series of tetraphenylethene derivatives, where a double bond is surrounded by phenyl groups. The restriction in intramolecular rotation accounts for making it AIE luminogen.4 Tian and co workers reported a group of triaryl-amine derivatives, which exhibit AIE.5 Park and co-workers have reported several material systems with AIE properties.6 Different mechanisms have been suggested to explain the AIE behavior, some of which include restriction of intramolecular rotation, formation of J-aggregates and intramolecular planarization.7The light-emitting property of a compound in solid state is dictated by the arrangement of the entire array of molecules rather than by an individual molecule. Studying crystal structures of the compounds provides essential information on how molecules are arranged with respect to their neighbors, what are the non covalent interactions binding them together and what are their conformational preferences are required to correlate the structure–property relationship. The knowledge of relating various properties with the supramolecular arrangement of the molecules in the solid state, the sensitivity of these properties with respect to the packing in the solids could lead to derive new strategies to tailor made the compounds according to our requirement. Supramolecular synthesis, which involves non covalent interaction has emerged as an efficient route to synthesize a wide range of solid forms. It is also a versatile approach to tune solid-state luminescence by controlling the molecular organization via intermolecular interactions. Draper and co workers have observed that the solid state luminescence of 2-cyano-3(4-(diphenylamino)phenyl)acrylic acid can be tuned by co-crystallization with substituted pyridines and amines, which results in effective control in the non covalent interactions.8 Recently, Varughese have highlighted the reports on non-covalent methods and their utility in attaining switchable solid state emitting molecular materials.9
In order to obtain structural correlations to solid state optoelectronic properties of molecular materials, it is necessary to obtain and analyze the crystal structure along with the spectroscopic results. In the present work, we have studied the solid state luminescence of Schiff base molecules and correlate the observed behavior with the crystal structure analysis. The solid state luminescence of a series of Schiff bases (Salen) involving alkyl chains was reported by Kawasaki et al. They have observed influence of alkyl chain length on their photo physical properties.10 The Schiff bases selected in the current work is shown in Scheme 1.11–17 The features selected for the Schiff bases include two pyridyl moities, an alkyl bridge and H or CH3 groups on the methinine carbon.
 | ||
| Scheme 1 Bis-pyridyl-alkyl-di-imines with alkyl spacers of different lengths. | ||
The compounds L1–L3 are reported to form wide range of coordination complexes of varied structural features and interesting properties. The compound L1a is explored extensively to prepare bimetallic and trimetallic coordination complexes of Cu(II), Cd(II), Ni(II), Ag(I) etc.11b–g and coordination polymers11h–l of diverse geometry and properties. The compound L1b has also shown to form coordination complexes with wide range of metals.12,13 The compounds L2a and L2b were shown to form mainly coordination complexes;14,15 while Hor and coworkers have reported a coordination polymer of L2b.15p Few coordination complexes of L3a and L3b are reported in the literature.16,17 Mitra and co workers have reported a heterometallic coordination complex of L3a involving Fe(II) and Cu(II) centers.16b Chandra and co workers have reported a coordination polymer of L3b with Mn(II),17e while rest of the reports of L3b involves coordination complexes of one or two metal centers.17
Although there are numerous reports on coordination complexes, the studies on the compounds L1–L3 itself, are very few. The solid state structure of L1a, L1b and L2a are reported by various research groups. The crystal structure of compound L1a is reported by Qing-Jin and co workers.11a while the structure of L1b is reported by Rudolph and co workers12e & also by Foxman and coworkers.12f Buyukgungor and coworkers14g have reported the crystal structure of L2a whereas no structural reports are there for the compounds L2b, L3a and L3b. The present work deals with the analysis of the structural features of the compounds L1a, L2a and L3b and correlating their photophysical properties observed in the solid state with that of their crystal structure. The photophysical properties of these compounds in solution is analyzed and compared with that obtained in the solid state.
Experimental section
General
Infra-red spectrum was recorded in FTIR ABB Bomen MB-3000. UV-vis absorption spectra were recorded in Shimadzu Spectrophotometer with model UV-2450. Fluorescence spectra were recorded in Shimadzu Spectrofluorophotometer with model RF-5301PC. Proton and carbon-13 nuclear magnetic resonance (1H NMR and 13C NMR) spectra were measured on a 400 MHz NMR spectrometer (Bruker).| Compound | L1a | L2a | L3b |
|---|---|---|---|
| a R1= Σ||Fo| − |Fc||/Σ|Fo|.b wR2 = [Σw(Fo2 − Fc2)2/Σw(Fo2)2]1/2, where w = 1/[σ2(Fo2) + (aP)2 + bP], P = (Fo2 + 2Fc2)/3. | |||
| Formula | C14 H14 N4 | C16 H18 N4 | C16 H18 N4 |
| Molecular weight | 238.29 | 266.34 | 266.34 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic |
| Space group | P21 | Pcab | P21/c |
| a/Å | 8.6694(4) | 19.379(5) | 9.2157(5) |
| b/Å | 17.8009(8) | 24.584(5) | 8.9944(5) |
| c/Å | 12.8064(6) | 12.300(5) | 9.3471(5) |
| α/° | 90.00 | 90.00 | 90.00 |
| β/° | 98.394(4) | 90.00 | 107.344(6) |
| γ/° | 90.00 | 90.00 | 90.00 |
| V/Å3 | 1955.15(16) | 5860(3) | 739.55(7) |
| Z | 6 | 16 | 2 |
| Dcalcd/g cm−3 | 1.214 | 1.208 | 1.196 |
| T/K | 293(2) | 293(2) | 293(2) |
| Theta (°) range for data used | 3.51 to 25.99 | 3.65 to 26.00 | 3.54 to 26.00 |
| Rint | 0.0418 | 0.0690 | 0.0441 |
| Reflections with I > 2σ(I) | 4636 | 2710 | 993 |
| No. of parameters refined | 493 | 366 | 91 |
| Final R (with I > 2σ(I)) | R1a = 0.0637; wR2b = 0.1609 | R1a = 0.0813; wR2b = 0.1901 | R1a = 0.0445; wR2b = 0.1085 |
| GOF on F2 | 1.001 | 1.038 | 1.042 |
Results and discussion
The structural descriptions of the compounds are studied to analyze its relation with the photophysical properties of the compounds.Structural description of L1a
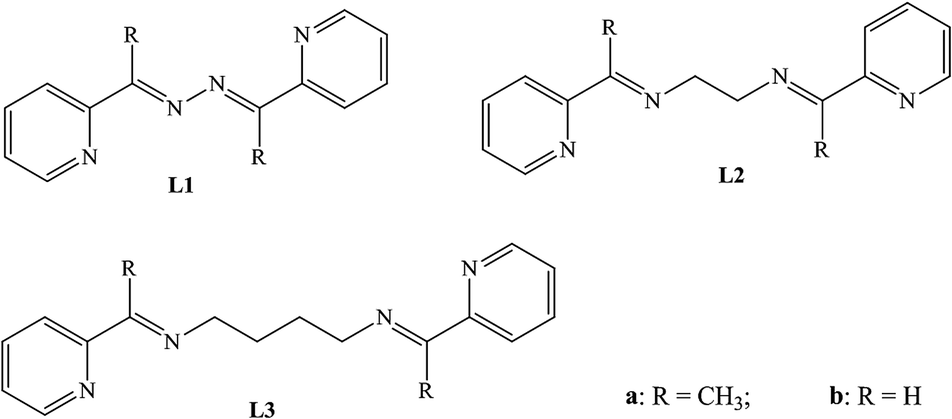
The crystal structure analysis of the compound L1a shows that it has crystallized in P21 and has three molecules in the asymmetric unit. The three molecules (molecule I, II and III) differ in the orientation of the pyridyl and imine moieties (Fig. 1a). On further analyzing the structure, it can be observed that the molecule I and molecule II are arranged in parallel manner; such that pyridyl rings and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N moieties of the two molecules are overlapping over one another. The distance between the CN of the two molecules are 3.906 and 3.943 Å, while the centroid-to-centroid distance between the two pyridyl rings are 3.888 Å and 4.009 Å.
N moieties of the two molecules are overlapping over one another. The distance between the CN of the two molecules are 3.906 and 3.943 Å, while the centroid-to-centroid distance between the two pyridyl rings are 3.888 Å and 4.009 Å.
 | ||
| Fig. 1 Illustration of the crystal structure of L1a: (a) asymmetric unit. The three molecules are labeled as molecule I, II & III; (b) non covalent interaction between the molecules; (c) packing of the molecules; (d) parallel arrangement of molecule I & II. | ||
Aromatic π⋯π interactions are present between the molecule I and molecule II. The non covalent interactions present in molecule III are rather significant. There are C–H⋯N and aromatic π⋯π interactions present between molecule III & molecule I and molecule III & molecule II (Fig. 1b). In fact, molecule III is sewing the molecule I and molecule II in a way such that they form dimers (Fig. 1c and d).
The crystal structure of a polymorph of L1a was reported by Qing-Jin and co workers where it has crystallized in P21/c space group. In that structure, there are three molecules in the asymmetric unit and the molecules adopt transoid geometry. In the present structure, the L1a molecules are crystallized in non centrosymmetric space group.
Structural description of L2a
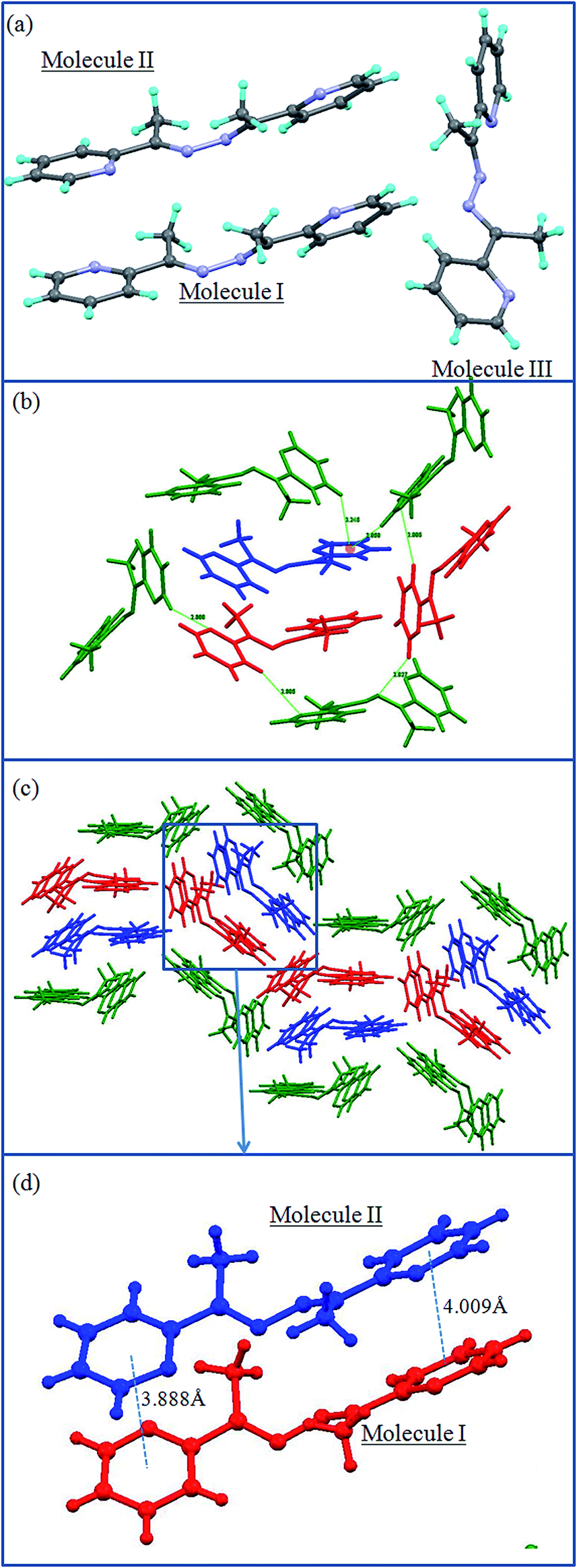
The crystal structure analysis of L2a shows that it has crystallizes in orthorhombic Pcab space group with two molecules present in the asymmetric unit (Fig. 2a).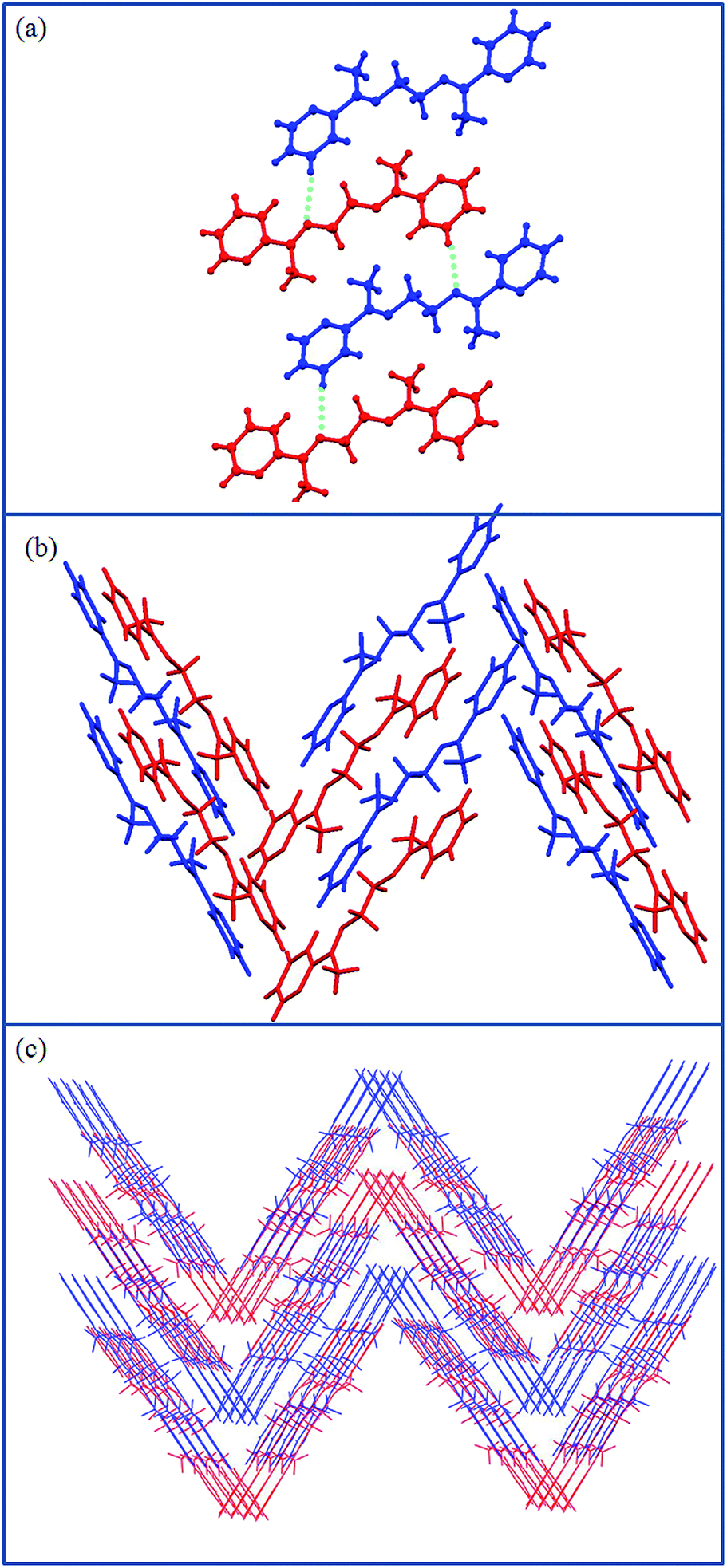 | ||
| Fig. 2 Illustration of the crystal structure of L2a: (a) asymmetric unit contains two molecules; the two types of molecules are shown in different colors; (b) aromatic interactions leading to zigzag layers; (c) packing of the zigzag layers. | ||
The two molecules differ in the orientations of the pyridyl and imine groups. The ethyl spacer of L2a is arranged in anti conformation, which gives it a linear geometry; flexible ethyl spacer, the arrangement of the molecules in the solid state give it a “rigid” planar-like geometry. The molecules are further assembled via aromatic interactions to form zigzag layers which are stacked further in 3D (Fig. 2b and c).
Structural description of L3b
The compound L3b crystallizes in monoclinic P21/c space group with half of the molecule present in the asymmetric unit (Fig. 3a). The molecules are held together by CH⋯N hydrogen bond interactions (2.781 Å; 3.709 Å; 176.02°) (Fig. 3b); which has resulted as the methyl group on the methinine carbon is replaced by hydrogen. The butyl chain in the spacer of L3b adopts gauche-anti-gauche conformation, which give “S”-shaped geometry to the molecule. The “S”-shaped molecules of L3b are further assembled via CH⋯N hydrogen bond to form corrugated layers. The corrugated layers are packed in an offset manner (Fig. 3c). The aromatic CH⋯π interactions are holding the layers together to pack them in 3D (Fig. 3d). | ||
| Fig. 3 Illustration of the crystal structure of L3b: (a) asymmetric unit: half molecule of L3b; (b) CH⋯N interactions leading to the formation of corrugated 2D layers; notice the gauche-anti-gauche conformation of butyl spacer of L3b; (c) offset packing of corrugated layers; (d) aromatic CH⋯π interactions between the L3b molecules of two adjacent layers. | ||
Powder XRD spectra of L1a, L2a and L3b
The powder XRD were taken for L1a, L2a and L3b (Fig. 4). The calculated PXRD of the three compounds were compared with that of the experimentally measured one in order to verify the phase purity of the compounds.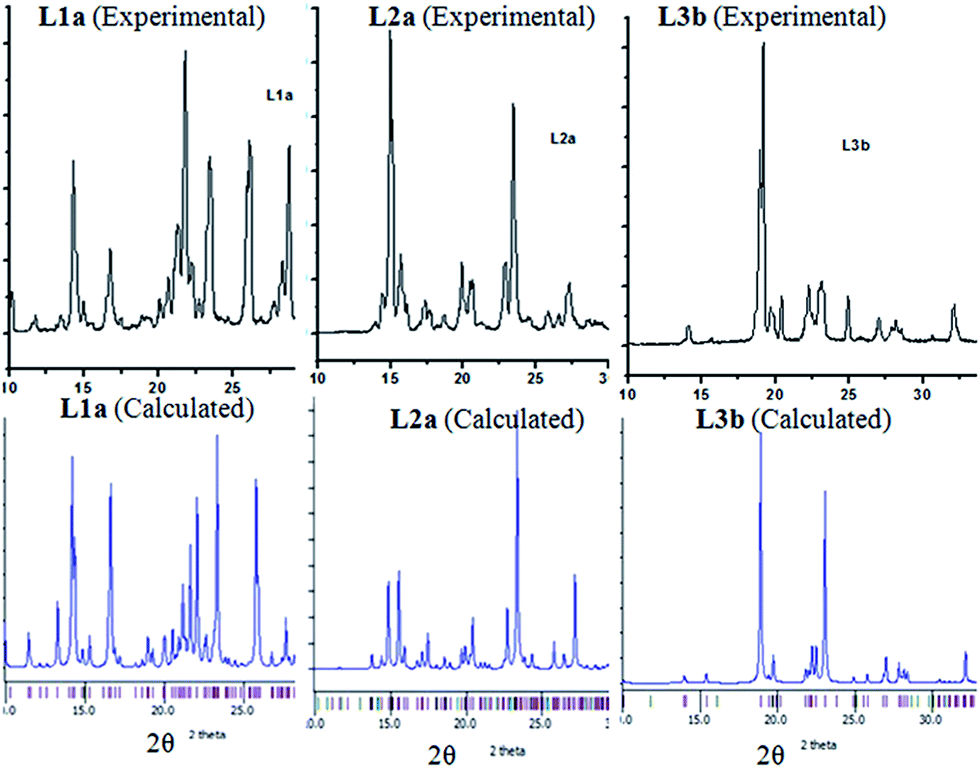 | ||
| Fig. 4 Experimentally measured and simulated powder XRD of L1a, L2a and L3b. | ||
UV-vis absorption spectra of the compounds
The UV absorption spectra were measured in CHCl3 for L1–L3. The compound L1a showed λmax at 286 nm and a hump at 259 nm; while for compound L1b, peak maxima were observed at 300 with a hump at 262 nm.The compounds L2a, L2b, L3a and L3b showed some similarity in the absorption spectra. All the four compounds showed two peaks at 240–241 and 270–273 nm, which may be attributed to π → π* and n → π* transitions, respectively (Fig. 5). The solid state UV-vis spectra of L1a, L2a and L3b are shown in Fig. S28–S30.†
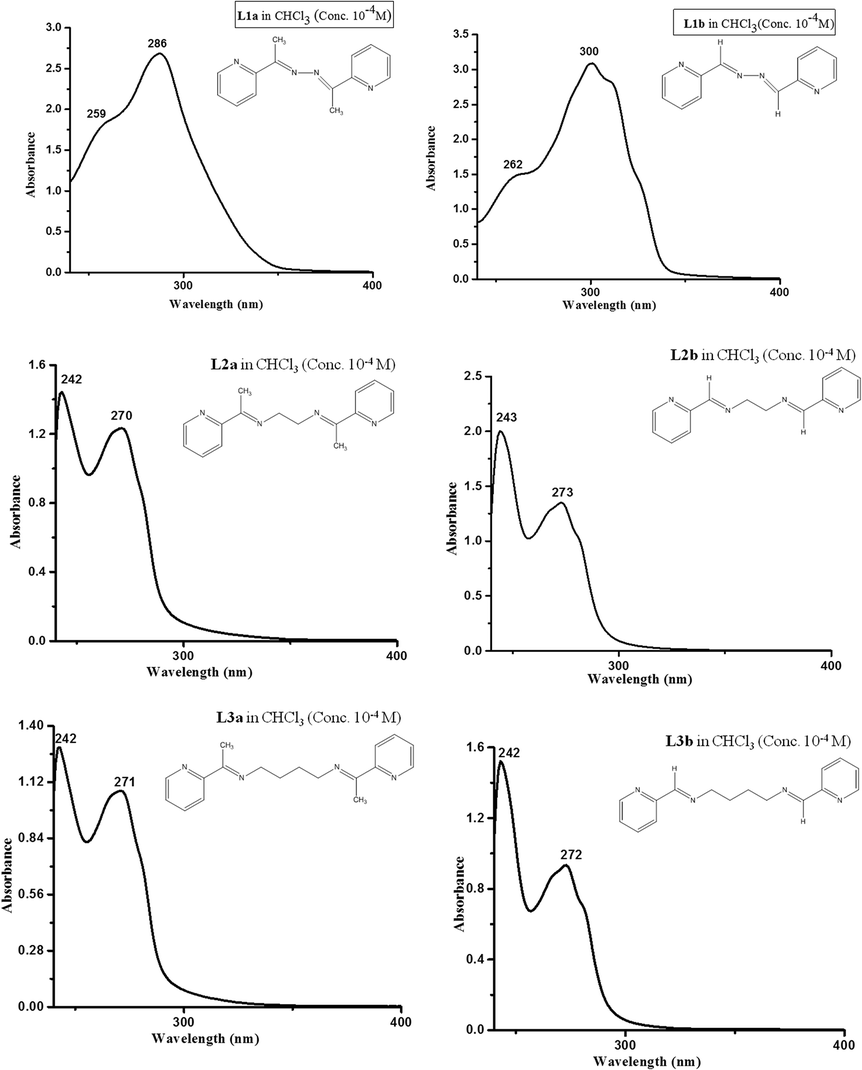 | ||
| Fig. 5 The UV absorption spectra of the compounds in CHCl3 (conc. of the compounds taken was 1 × 10−4 M). | ||
The chromophore 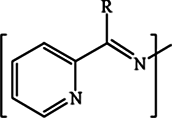 is responsible for both the transitions in compounds L2a, L2b, L3a and L3b, while in L1a and L1b, the extended conjugation throughout the molecule shifts the peak to longer wavelength.
is responsible for both the transitions in compounds L2a, L2b, L3a and L3b, while in L1a and L1b, the extended conjugation throughout the molecule shifts the peak to longer wavelength.
Excitation spectra and photoluminiscence spectra of L1a and L1b
The excitation and photoluminescence spectra were measured for L1a in CHCl3. At 10−5 M concentration, L1a showed excitation maxima at 315 nm, when the emission wavelength was kept at 365 nm (Fig. 6).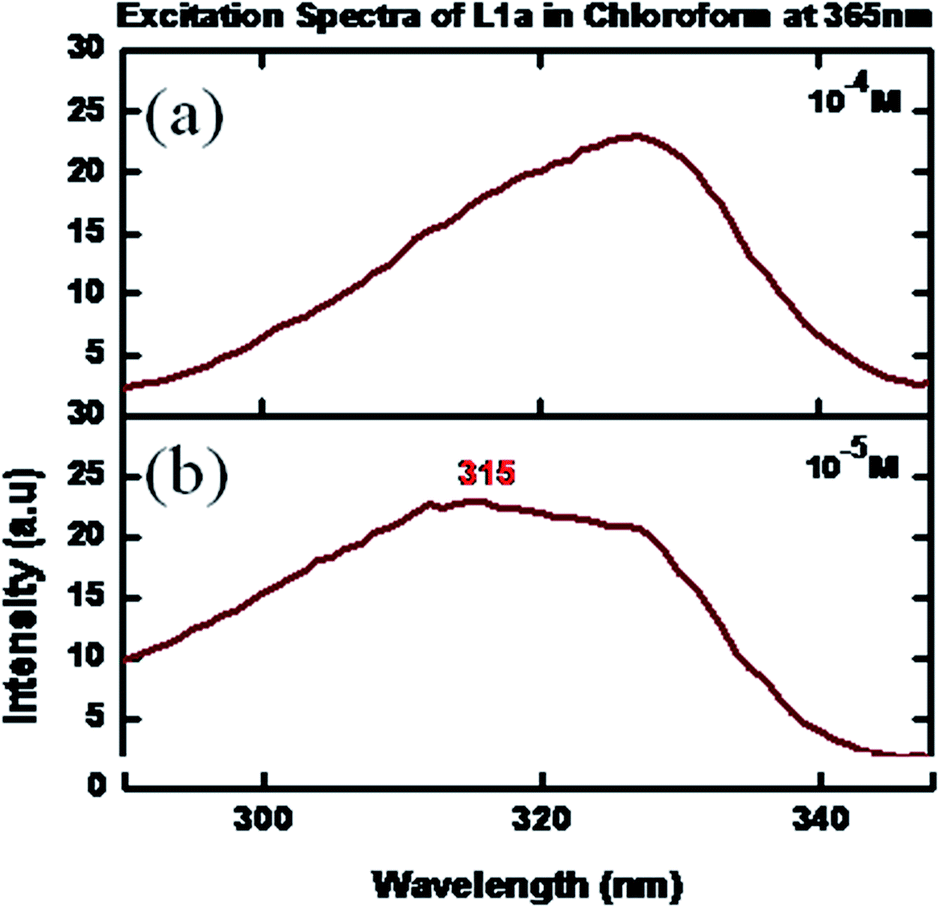 | ||
| Fig. 6 Excitation spectra of L1a in CHCl3 in different concentrations: (a) 10−4 M, (b) 10−5 M. | ||
The photoluminescence spectra at a concentration of 10−6 M showed a peak at 365 nm, when an excitation wavelength of 315 nm was used. Quenching of photoluminescence was observed as the concentration was increased (Fig. 7). The solid state photoluminescence was negligible (peak at λ = 535 nm) with a very low quantum yield (Fig. 8). The excitation and PL spectra of L1b also show similar behavior (Fig. S31 and S32†).
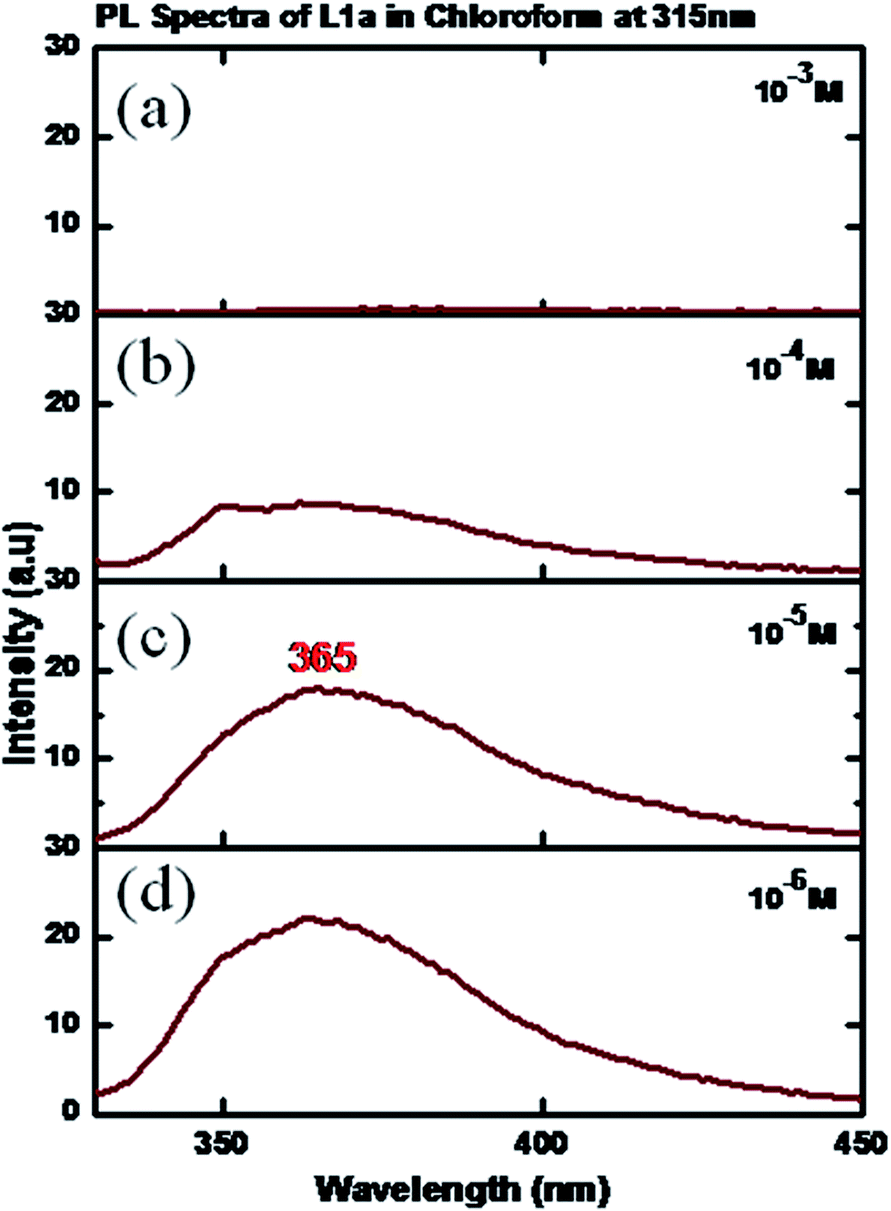 | ||
| Fig. 7 PL spectra of L1a in CHCl3 in different concentrations: (a) 10−3 M, (b) 10−4 M, (c) 10−5 M, (d) 10−6 M. | ||
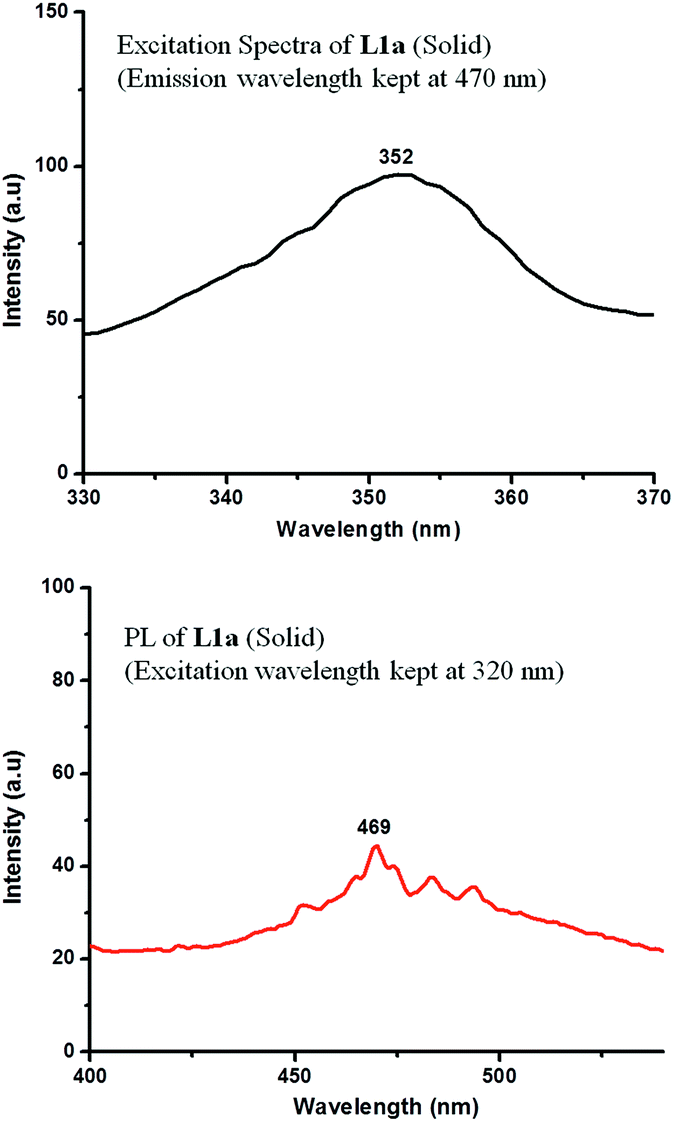 | ||
| Fig. 8 Excitation spectra and PL spectra of L1a in solid state. | ||
Tang et al. have reported Aggregation Induced Enhanced Emission (AIEE) for salicylaldehyde azine derivatives; which has two salicylaldimine moieties connected by rotatable N–N single bond (Scheme 2).20 They have suggested that intramolecular hydrogen bonds of salicylaldimine moieties and stacking of molecules resulted in AIEE; which resulted due to the inhibition of free intramolecular rotation when changed from solution to aggregate state. Though the skeletal structure of L1a and L1b is similar to salicylaldehyde azines, but in L1a and L1b quenching of photoluminescence is observed on moving from “free state” to aggregated state. The crystal structure of L1a showed some important aspects about the packing of the molecules, which is associated with quenching of fluorescence of L1a in solid state. The arrangement of the molecules of L1a in a π⋯π stacked molecular pairs (dimers) facilitates the two molecules forming an excimer, which explains its non fluorescent behavior in the solid state (Fig. 1d).21
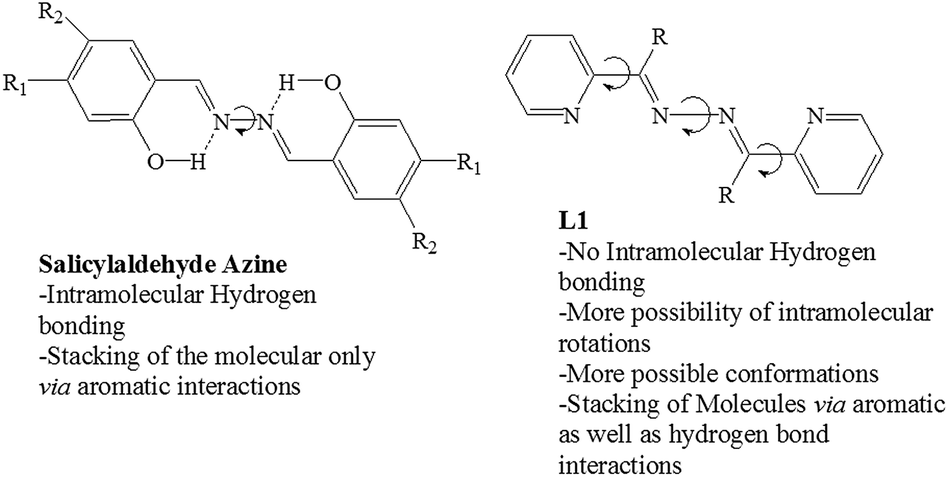 | ||
| Scheme 2 Comparison of L1 with salicyladehede azines.20 | ||
Excitation spectra and photoluminescence spectra of L2a and L3a
The excitation spectra of L2a were taken in CHCl3 by varying the concentration. The spectra showed maxima at a wavelength of 370–385 nm, when the emission wavelength was kept at 455 nm.Increasing the concentration of L2a resulted in increase in the intensity along with a slight red shift of the peak maxima (Fig. 9). The excitation spectra of L2a at an emission wavelength of 370 nm are shown in Fig. S33.†
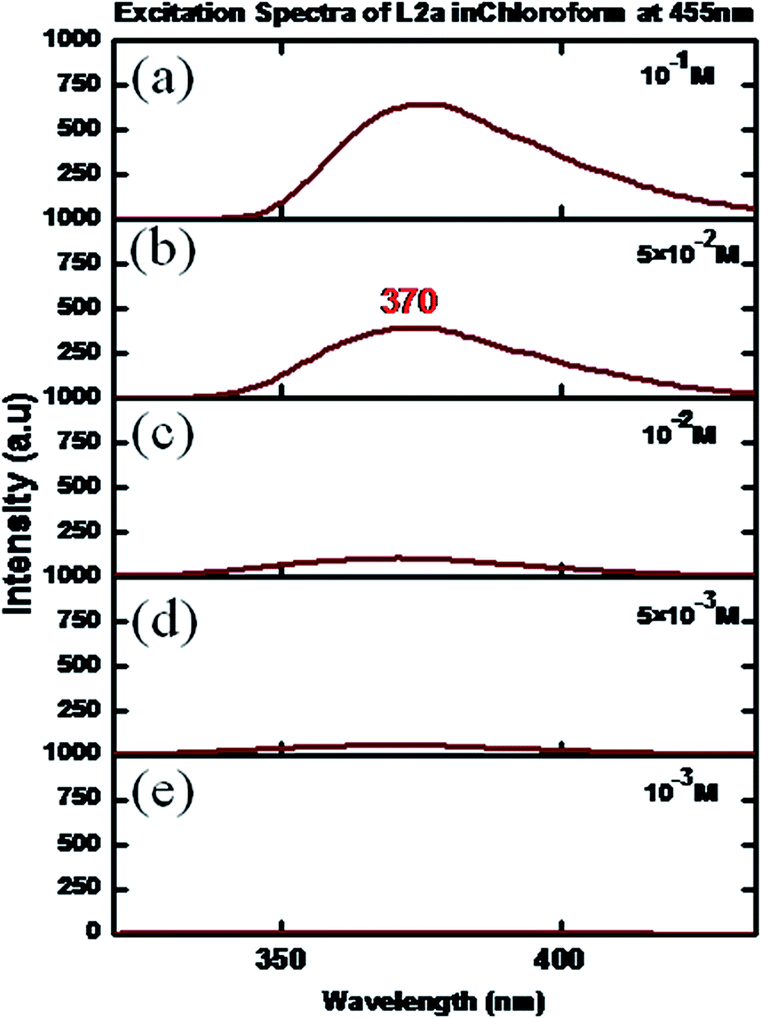 | ||
| Fig. 9 Excitation spectra of L2a in CHCl3 in different concentrations: (a) 10−1 M, (b) 5 × 10−2 M (c) 10−2 M (d) 5 × 10−3 M, (e) 10−3 M. | ||
The photoluminescence spectra of L2a in CHCl3 showed a weakly intense peak at 455 nm (when an excitation wavelength of 370 nm was used) at a concentration of 10−3 M. The change in concentration resulted in change in intensity of the peak; slight shift was observed in the peak position (Fig. 10). The solid state photoluminescence spectra of L2a shows highly intense band at 453 nm (Fig. 11). The enhancement of the luminescence intensity along with a red shift of peak positions for the solid L2a is related to crystallization induced enhanced mission.
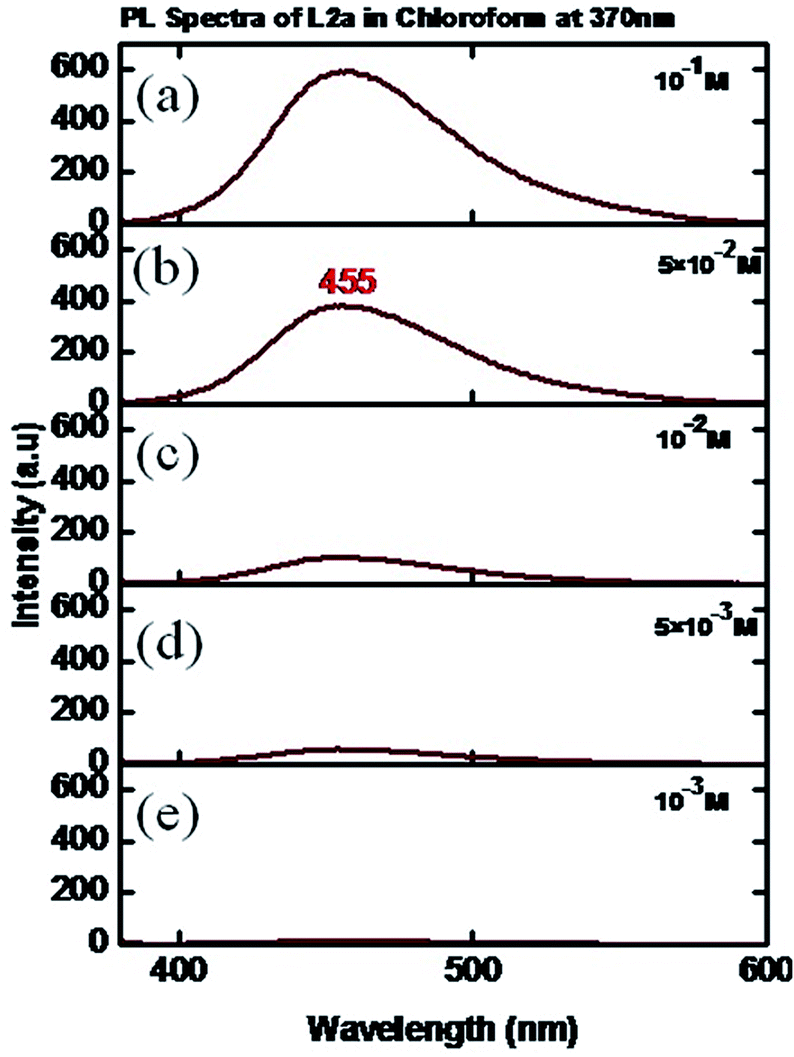 | ||
| Fig. 10 PL spectra of L2a in CHCl3 in different concentrations: (a) 10−1 M, (b) 5 × 10−2 M (c) 10−2 M (d) 5 × 10−3 M, (e) 10−3 M. | ||
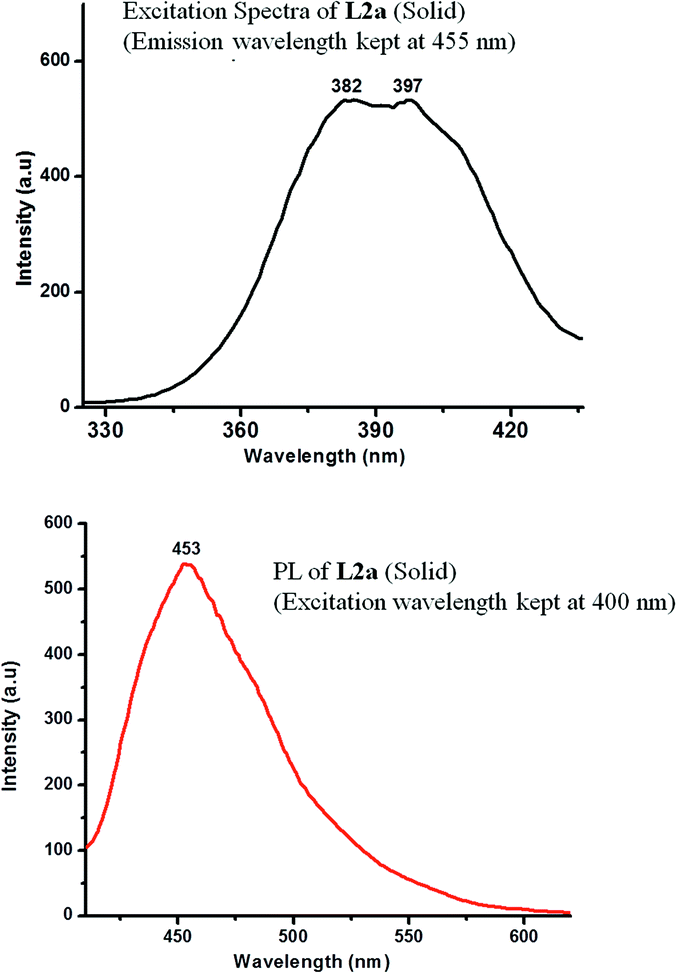 | ||
| Fig. 11 Excitation spectra (emission was kept at 455 nm) and PL spectra (at an excitation wavelength of 400 nm) of L2a in solid state. | ||
The crystal structure of L2a showed that although the spacer has a flexible ethyl chain, the arrangement of the molecules in the solid state give it a “rigid” planar-like geometry (Fig. 2). The enhanced emission in the solid state of L2a can be interpreted in terms of simultaneous effects of intra- and intermolecular effects exerted by the aggregation of the molecules. The planar-like geometry of the molecule in the solid state activates the radiation process, while the aggregation morphology resulting due to the supramolecular arrangement of the molecules also influences the emission process. The molecules in L2a are arranged in parallel manner but they are arranged in a specific slipped-type pattern, so that the excimer formation is avoided (Fig. 2b). The presence of the ethyl spacer in L2a made it possible to have a slipped-type arrangement of the molecules. This type of arrangement in L1 is not possible due to steric reasons and packing of the molecules is mostly via aromatic π⋯π interactions, which promotes the formation of dimers (Scheme 3).
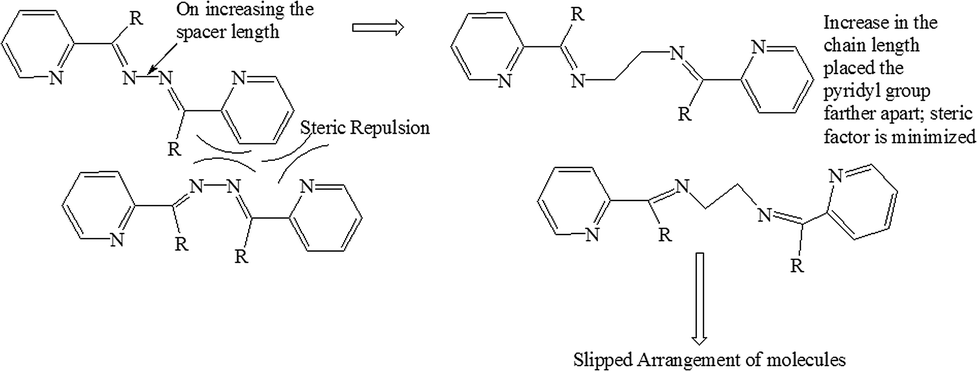 | ||
| Scheme 3 Comparison of L1 and L2 to understand the effect of spacer length on slipped-type arrangement of the molecules. | ||
The excitation and the PL spectra were recorded for L3a, which showed a similar behavior as L2a. At 10−3 M concentration, excitation spectra of L3a in CHCl3 showed a peak at 370 nm (when the emission wavelength is kept at 460 nm). On increasing the concentration, the slight red shift of peak maxima along with increase in the intensity was observed (Fig. S34 and S35†).
The PL spectra in CHCl3 at an excitation wavelength of 450 nm showed enhancement in the intensity with increase in concentration and a red shift by ∼10 nm of the peak maxima. The solid state spectra of L3a showed a highly intense band at 495 nm (Fig. S36†). The similarity in excitation and the PL spectra of L2a and L3a suggests that the packing of the molecules in L3a may be similar to L2a, where all anti conformation of the butyl spacer results in slipped type arrangement of the molecules and diminishes the possibility of forming excimers (Scheme 4). The crystal structure analysis of L3a couldn't be done because the good quality crystals of L3a couldn't be obtained.
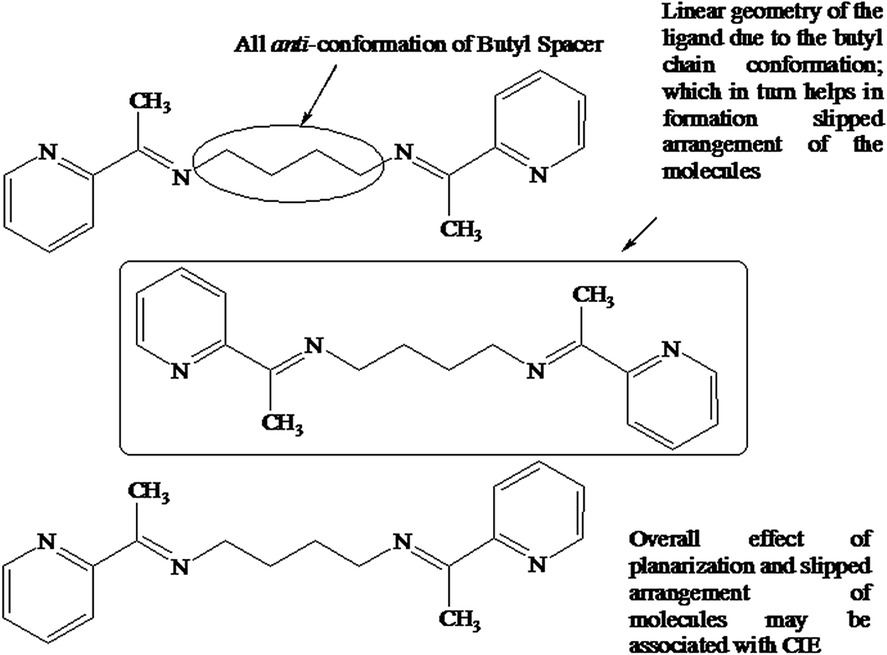 | ||
| Scheme 4 Suggested structure of L3a in the solid state to account for the CIE. | ||
Excitation spectra and photoluminescence spectra of L2b and L3b
Interesting photoluminescence spectral properties were observed for L2b and L3b, where the methyl group on the methinine carbon is replaced by H-atom, so that there will be enough opportunity for the formation of non-covalent interactions. The concentration dependent excitation spectra of L2b in CHCl3 showed that at a concentration of 10−3 M, the spectra had maxima at 365 and 420 nm (emission wavelength was kept at 460 nm). On increasing the concentration, the intensity of the peak at 420 nm was increased, while a hump appeared at 365 nm. The peak intensity at 365 nm also increases with concentration (Fig. 12).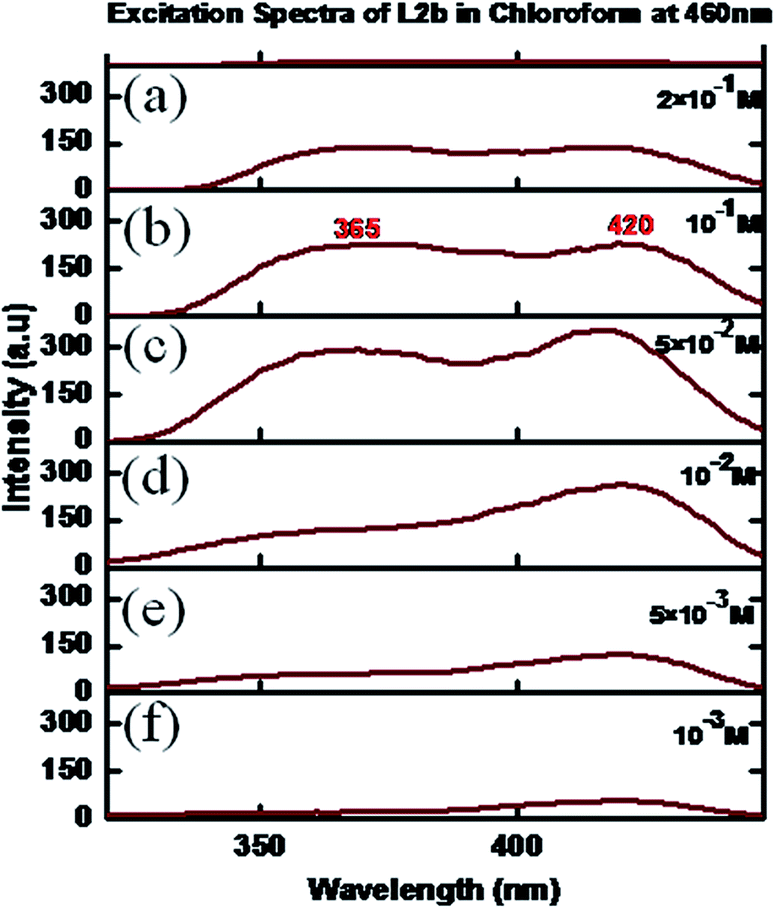 | ||
| Fig. 12 Excitation spectra of L2b in CHCl3 in different concentrations: (a) 2 × 10−1 M, (b) 10−1 M (c) 5 × 10−2 M (d) 10−2 M (e) 5 × 10−3 M, (f) 10−3 M. | ||
The concentration dependent PL spectra of L2b in CHCl3 were measured at an excitation wavelength of 420 nm. A peak at 465 nm was observed at a concentration of 10−3 M, which on further increasing concentration resulted in increase in the peak intensity. But at a concentration of 5 × 10−2 M, a hump appeared at 545 nm, which became intense with the concentration increase. Finally, at a concentration of 5 × 10−1 M, the peak at 455 nm disappeared, while an intense peak at 555 nm was observed (Fig. 13). In the solid state spectra of L2b, strong emission peak appeared in the region between 555 nm (Fig. 14).
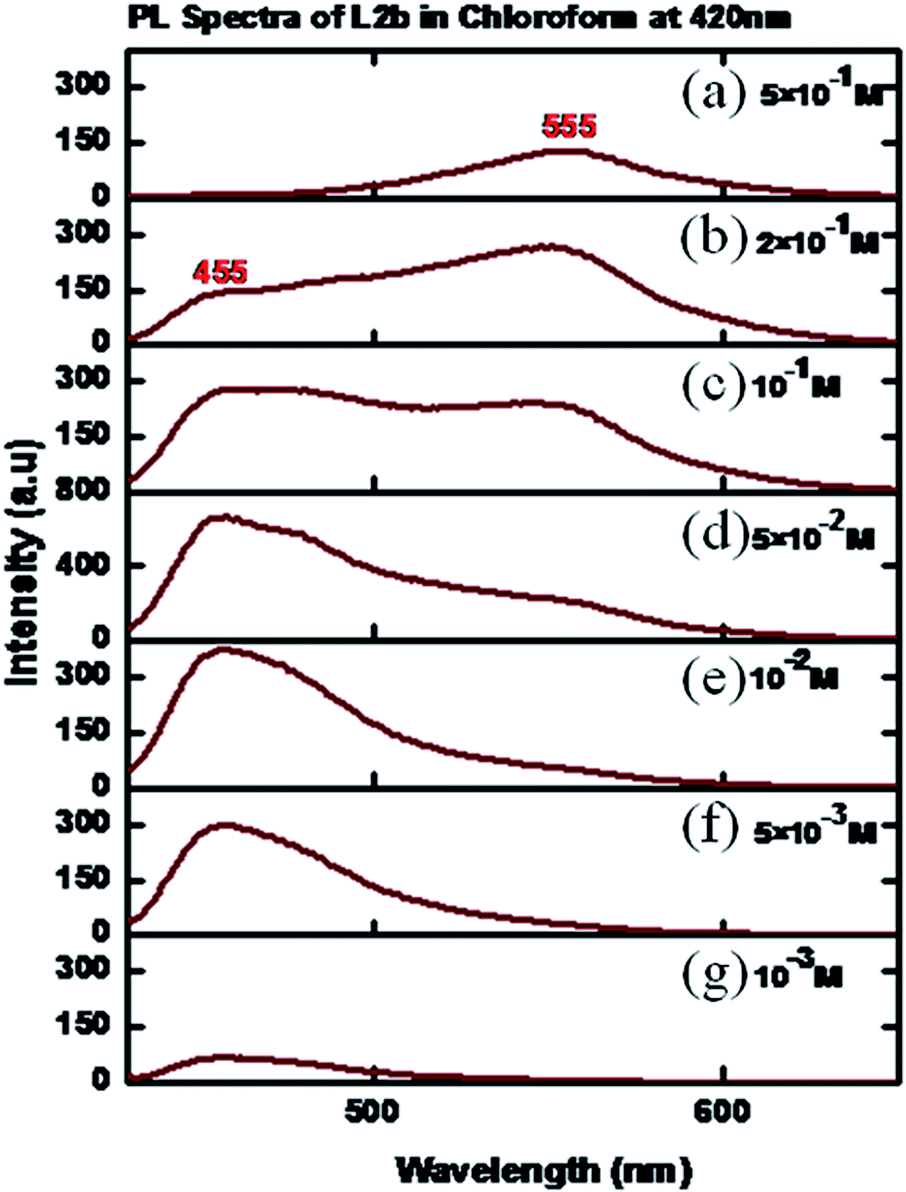 | ||
| Fig. 13 PL spectra of L2b in CHCl3 in different concentrations (a) 5 × 10−1 M; (b) 2 × 10−1 M; (c) 10−1 M (d) 5 × 10−2 M; (e) 10−2 M (f) 5 × 10−3 M, (g) 10−3 M. | ||
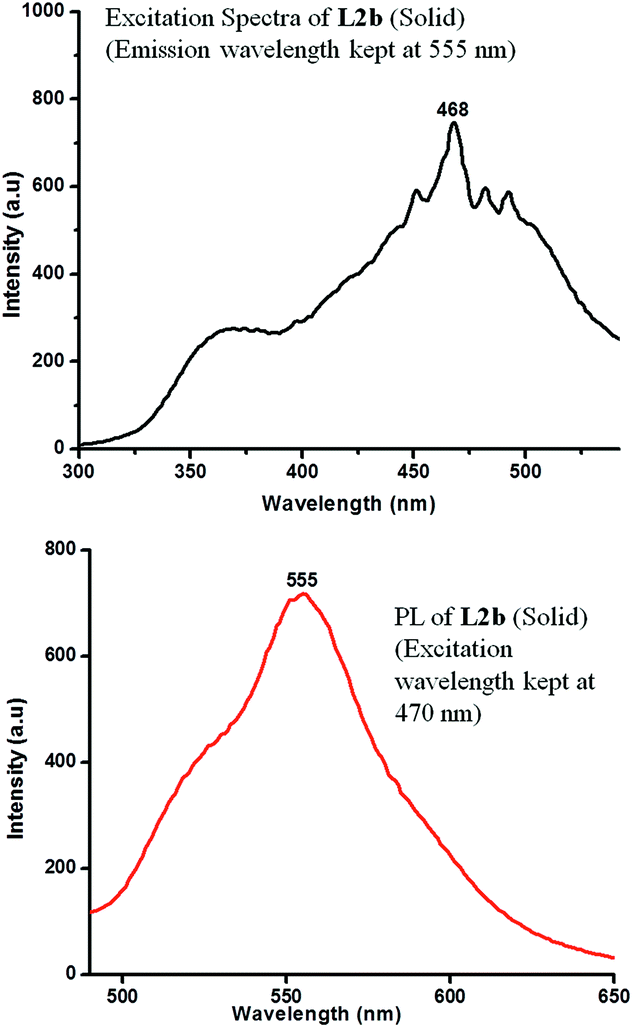 | ||
| Fig. 14 Excitation spectra (emission was kept at 555 nm) and PL spectra (at an excitation wavelength of 470 nm) of L2b in solid state. | ||
The excitation and photoluminescence spectra of L3b in CHCl3 showed similar behaviour as that of L2b. The concentration dependent excitation spectra of L3b in CHCl3 showed that at a concentration of 5 × 10−3 M, the spectra had maxima at 365 and 420 nm (emission wavelength was kept at 460 nm) (Fig. 15).
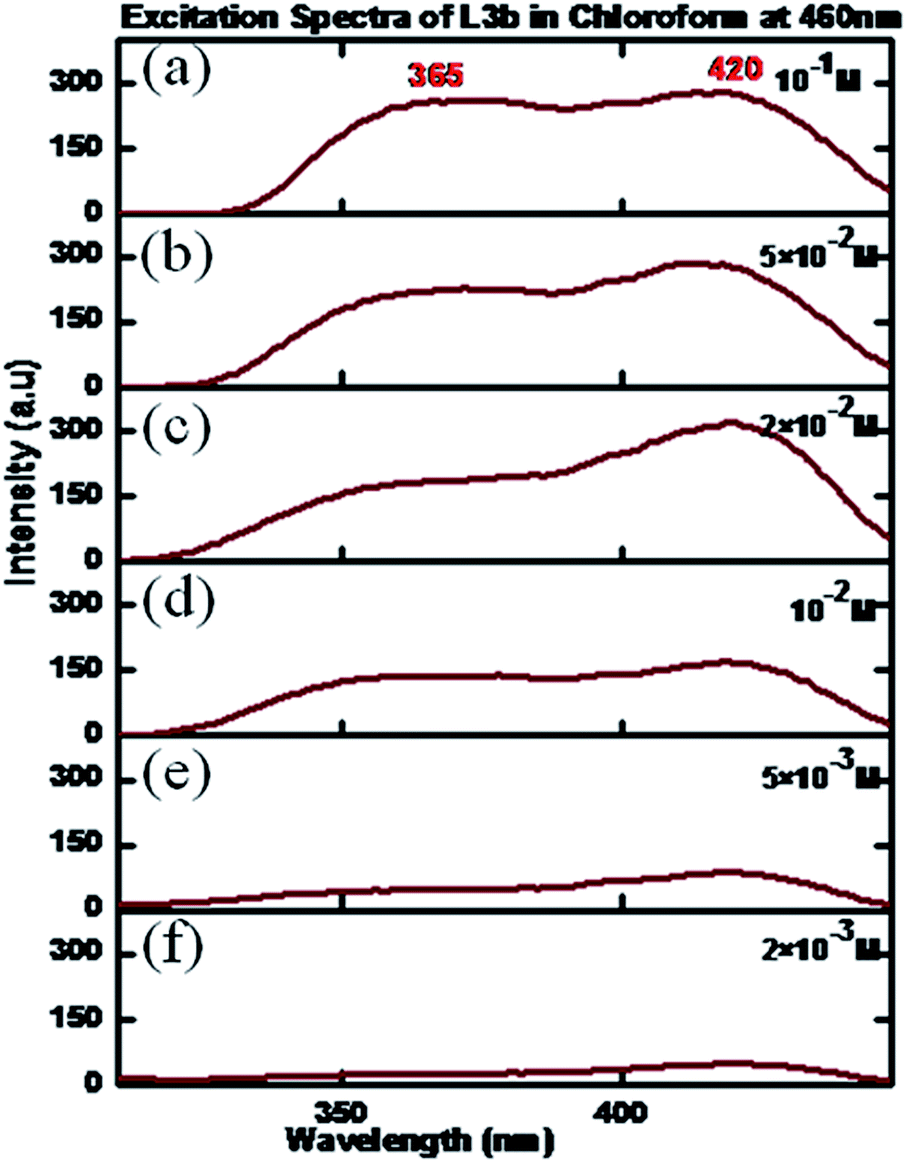 | ||
| Fig. 15 Excitation spectra of L3b in CHCl3 in different concentrations: (a) 10−1 M, (b) 5 × 10−2 M, (c) 2 × 10−2 M (d) 10−2 M (e) 5 × 10−3 M, (f) 10−3 M. | ||
The PL spectra of L3b for a concentration of 5 × 10−3 M showed maxima at 460 nm, when an excitation wavelength of 420 nm was used. On increasing the concentration, the increase in the intensity of peak maxima was observed. However, at a concentration of 5 × 10−2 M, a hump appeared at 545 nm, intensity of which increases with increase in the concentration. Finally, at a concentration of 5 × 10−1 M, an intense peak was present at 545 nm while the peak at 460 nm disappeared (Fig. 16). The solid-state photoluminescence spectra of L3b showed strong emission peak appeared in the region between 548 nm (Fig. 17).
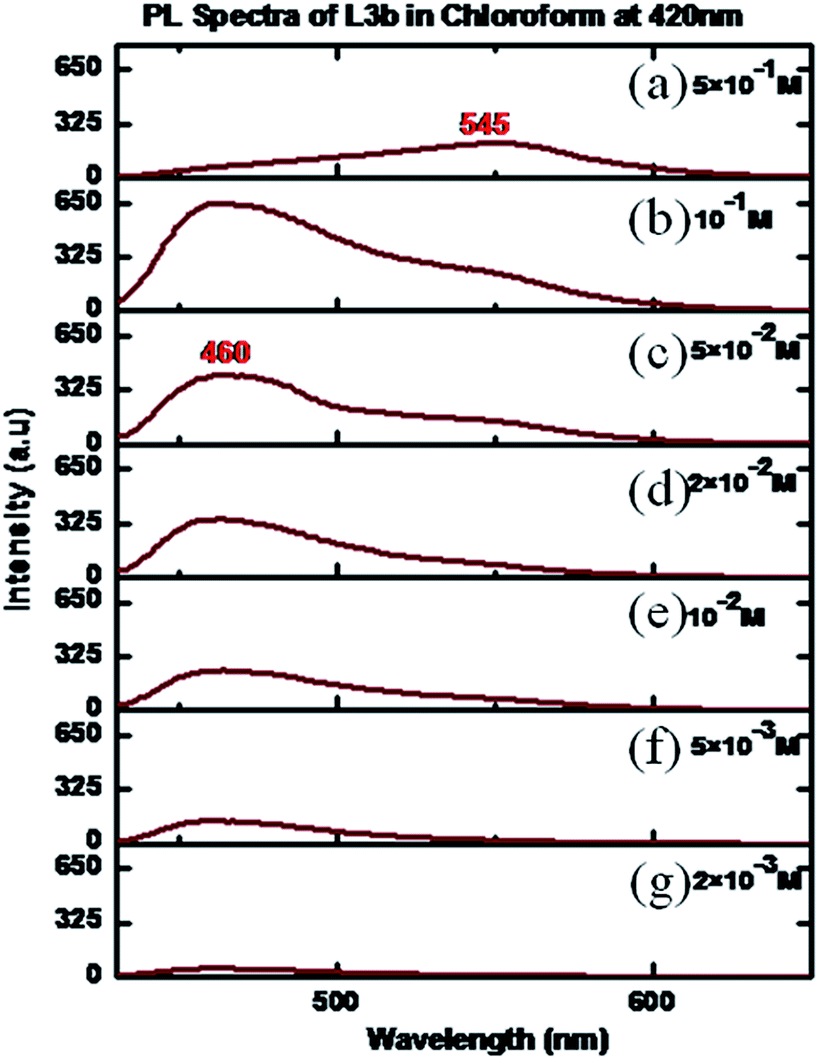 | ||
| Fig. 16 PL spectra of L3b in CHCl3 in different concentrations (a) 5 × 10−1 M; (b) 10−1 M; (c) 5 × 10−2 M; (d) 2 × 10−2 M; (e) 10−2 M (f) 5 × 10−3 M; (g) 2 × 10−3 M. | ||
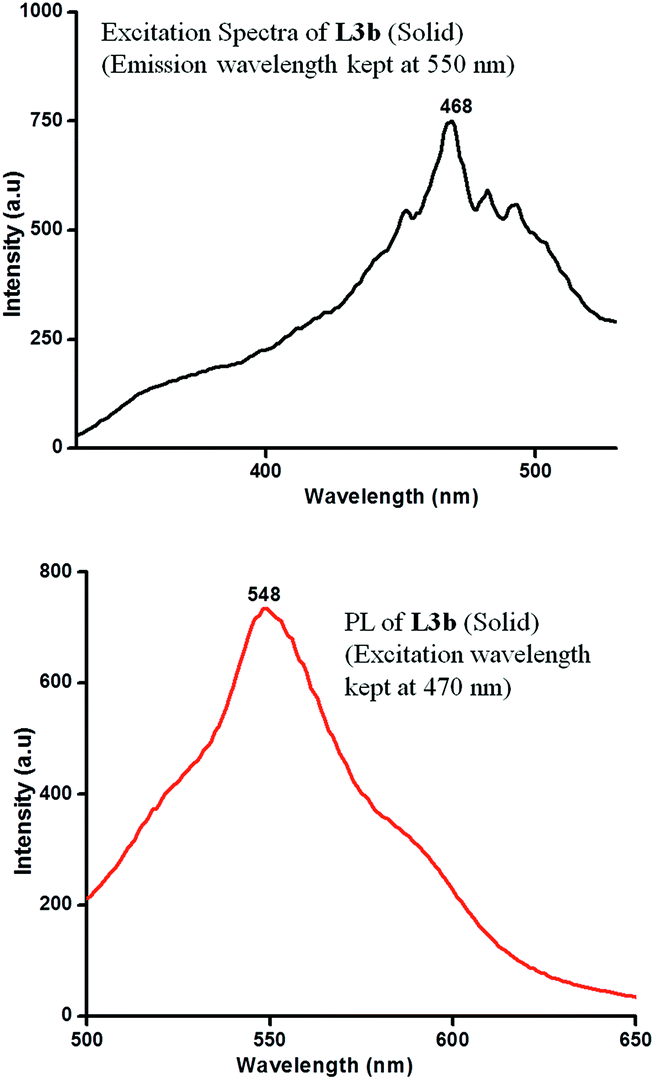 | ||
| Fig. 17 Excitation spectra (emission was kept at 550 nm) and PL spectra (at an excitation wavelength of 470 nm) of L3b in solid state. | ||
The appearance of a new peak at a concentration of 5 × 10−1 M followed by a red shift of the same peak in case of solid L2b and L3b suggested the formation of non-covalently bonded “species” of the molecules. The large bathochromic shift of the emission peak suggested the formation of chromophore dimers of L2b and L3b in the aggregated state (Scheme 5).
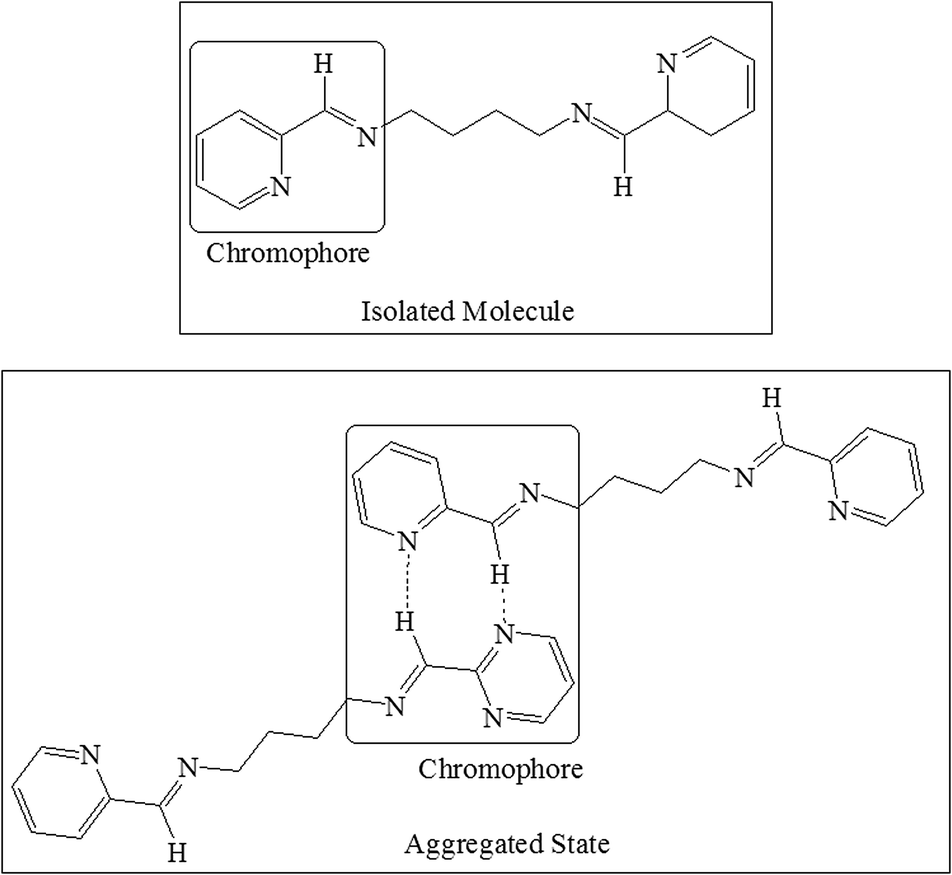 | ||
| Scheme 5 Hydrogen bonded dimeric species in the aggregated state of L2b and L3b; which account for appearance of new peak in high concentrated solutions and a large bathochromic shift in case of solid state PL spectra. | ||
NMR assay for detecting the aggregation in compound L3b
The expected nature of the NMR spectra for non-aggregating compound includes sharp NMR resonance peaks at all concentrations and peak position is independent of the concentration. In case of aggregating compounds, higher concentrations result in self association of the molecules, while dilution will result in free state of the molecules. As a result, the effect of concentration changes will be observed in the NMR spectra. The NMR features that may become concentration dependent for aggregating compounds include chemical shifts, shape and intensity of the peaks.22The 1H NMR spectra of L3b was recorded as a function of compound concentration (Fig. 18 and S37†). The protons in the aromatic region show change in chemical shift when the concentration of L3b in CDCl3 was increased from 10−2 M to 1 M. The crystal structure of L3b shows the packing of the molecules, where the imine proton (He in the Fig. 18) is in close proximity with pyridyl nitrogen. The protons Ha, Hb and Hc interact with pyridyl ring of another molecule as shown in Fig. 3d. The aromatic protons and the imine proton are becoming more shielded protons on increasing the concentration of L3b.
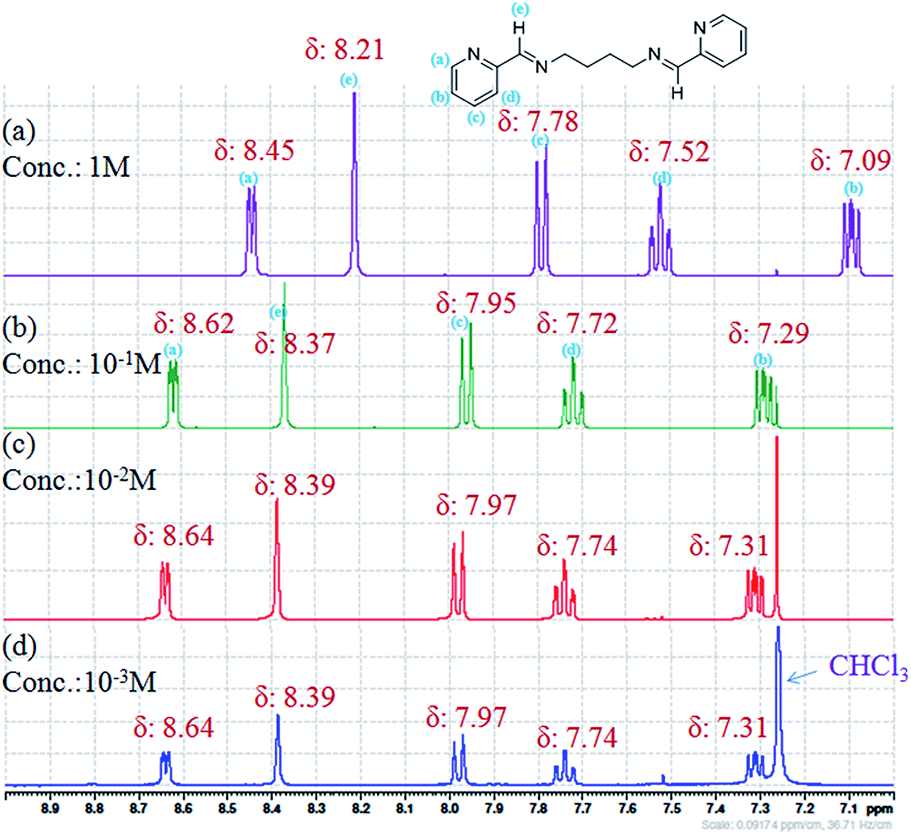 | ||
| Fig. 18 1H NMR spectra of L3b in CDCl3 by changing the concentration of compound; (a) 1 M; (b) 10−1 M; (c) 10−2 M; (d) 10−3 M. | ||
The NOESY of L3b was recorded to further observe the aggregation of the molecules at high concentration (Fig. 19, S38 and S39). The spectra recorded at a concentration of 1 M of L3b in CDCl3 shows interaction between the aromatic and the aliphatic protons (Fig. 19a), while very less interaction is observed at a concentration of 10−2 M. Further, quite intense interaction between the aromatic protons is observed at a higher concentration than that at a lower concentration.
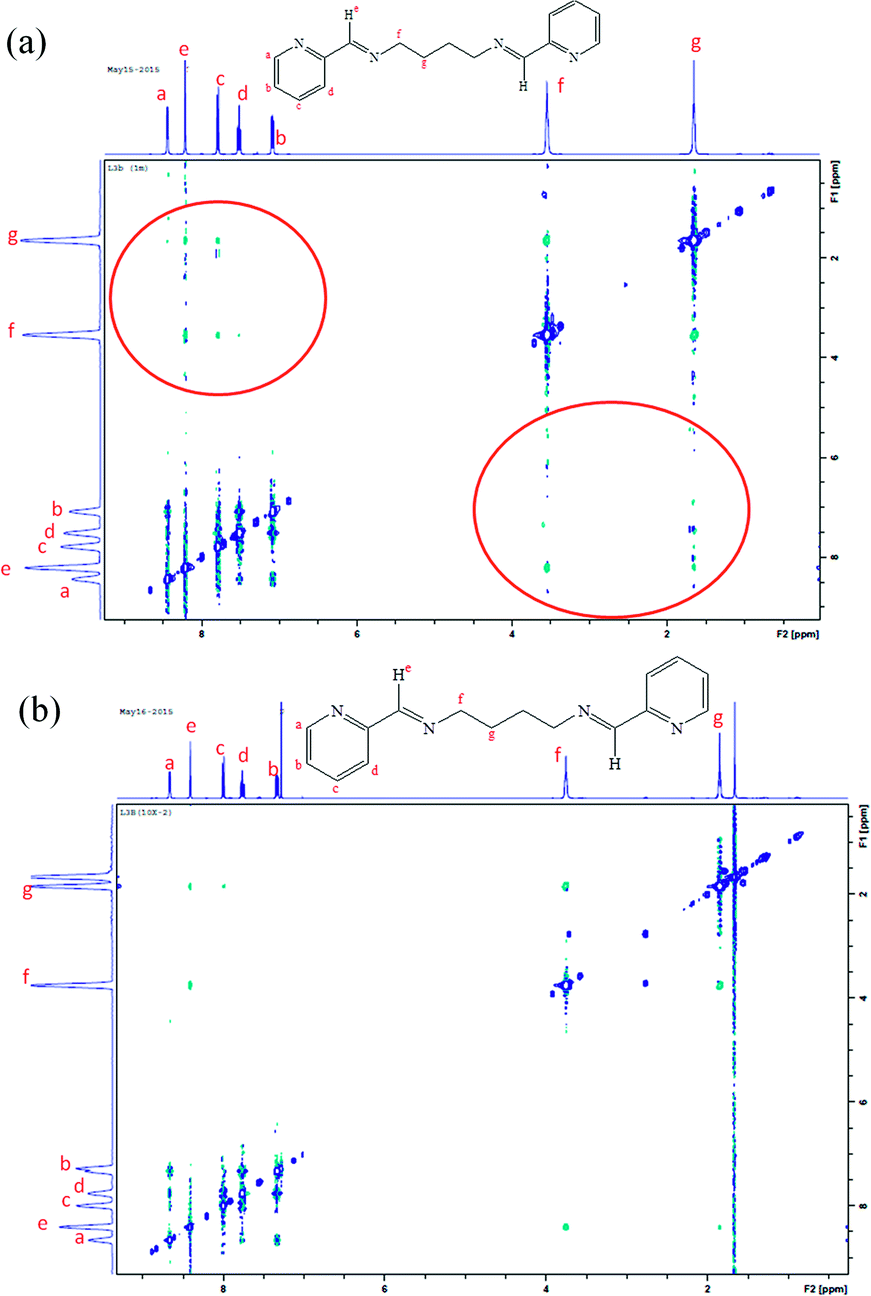 | ||
| Fig. 19 NOESY of L3b in CDCl3 at a concentration of (a) 1 M; (b) 10−2 M. | ||
Conclusions
The study of structure and photophysical property has revealed some important rationale regarding the interplay of molecular geometry and non-covalent interactions in deciding the light emitting properties of compounds. The compounds L1a and L1b show PL spectra in dilute solution and quenching in PL spectra is observed on increasing concentration. The compounds L2a, L2b, L3a and L3b, show enhancement in the PL intensity on increasing the concentration.Although the molecule L1a and L1b are rigid, when compared to L2a, L2b, L3a and L3b, doesn't show any emission spectra in the solid state, while L2a, L2b, L3a and L3b show highly intense PL spectra in the solid state.
The flexible spacer (alkyl bridge) in L2a, L2b, L3a and L3b, helped in assembling the molecules in a way which results in enhancement in PL spectra on aggregation. In L2b and L3b, the presence of hydrogen atom in place of CH3-group further enhances the possibility of forming various aggregates/non covalently bonded species which enhances the emission intensity.
Acknowledgements
We gratefully acknowledge the financial support from the Seed Grant Scheme-2011 of BITS Pilani & DST (SR/FT/CS-24/2011) and Instrumentation facility from the DST FIST & UGC-SAP to the Department of Chemistry, BITS Pilani, Pilani Campus. R.K. acknowledges financial support received under DST mega research project (SR/S2/CMP-47/2003). F.B. thanks DST for providing research fellowship. We also acknowledge the NMR facility of the Department of Chemistry, BITS Pilani, Pilani Campus and Mr Shiv Dhiman for recording the concentration dependent NMR and NOESY spectra.Notes and references
- (a) A. C. Grimsdale and K. Müllen, Adv. Polym. Sci., 2008, 212, 1 CAS; (b) Y. Zhang, B. Liu and Y. Cao, Chem.–Asian J., 2008, 3, 739 CrossRef CAS PubMed; (c) J. Chen and Y. Cao, Macromol. Rapid Commun., 2007, 28, 1714 CrossRef CAS PubMed; (d) V. W. W. Yam and K. M. C. Wong, Top. Curr. Chem., 2005, 257, 1 CAS; (e) F. J. M. Hoeben, P. Jonkheijm, E. W. Meijer and A. P. H. J. Schenning, Chem. Rev., 2005, 105, 1491 CrossRef CAS PubMed; (f) W. Lu, M. C. W. Chan, N. Y. Zhu, C. M. Che, Z. He and K. Y. Wong, Chem.–Eur. J., 2003, 9, 6155 CrossRef CAS PubMed; (g) U. H. F. Bunz, Chem. Rev., 2000, 100, 1605 CrossRef CAS PubMed; (h) F. Hide, M. A. Diaz-Garcia, B. J. Schwartz and A. J. Heeger, Acc. Chem. Res., 1997, 30, 430 CrossRef CAS; (i) J. H. Burroughes, D. D. C. Bradley, A. R. Brown, R. N. Marks, K. Mackay, R. H. Friend, P. L. Burns and A. B. Holmes, Nature, 1990, 347, 539 CrossRef CAS PubMed; (j) C. W. Tang and S. A. Vanslyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS PubMed.
- (a) H. Wohltjen and A. W. Snow, Anal. Chem., 1998, 70, 2856 CrossRef CAS; (b) A. N. Shipway, E. Katz and I. Willner, ChemPhysChem, 2000, 1, 18 CrossRef CAS; (c) A. Pucci, F. D. Cuia, F. Signori and G. Ruggeri, J. Mater. Chem., 2007, 17, 783 RSC; (d) Z. Ning, Z. Chen, Q. Zhang, Y. Yan, S. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799 CrossRef CAS PubMed; (e) M. Kinami, B. R. Crenshaw and C. Weder, Chem. Mater., 2006, 18, 946 CrossRef CAS; (f) A. Hagfeldt and M. Grätzel, Chem. Rev., 1995, 95, 49 CrossRef CAS; (g) M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 2013 CrossRef CAS; (h) X. Michalet, F. Pinaud, T. D. Lacoste, M. Dahan, M. P. Bruchez, A. P. Alivisatos and S. Weiss, Single Mol., 2001, 2, 261 CrossRef CAS.
- (a) J. Luo, Z. Xie, J. W. Y. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu, D. Zhu and B. Z. Tang, Chem. Commun., 2001, 1740 RSC; (b) B. Z. Tang, X. Zhan, G. Yu, P. P. S. Lee, Y. Liu and D. Zhu, J. Mater. Chem., 2001, 11, 2974 RSC.
- Q. Zeng, Z. Li, Y. Dong, C. Di, A. Qin, Y. Hong, L. Ji, Z. Zhu, C. K. W. Jim, G. Yu, Q. Li, Z. Li, Y. Liu, J. Qin and B. Z. Tang, Chem. Commun., 2007, 70 RSC.
- Z. J. Ning, Z. Chen, Q. Zhang, Y. L. Yan, S. X. Qian, Y. Cao and H. Tian, Adv. Funct. Mater., 2007, 17, 3799 CrossRef CAS PubMed.
- B. K. An, S. K. Kwon, S. D. Jung and S. Y. Park, J. Am. Chem. Soc., 2002, 124, 14410 CrossRef CAS PubMed.
- (a) Y. Hong, J. W. Y. Lama and B. Z. Tang, Chem. Commun., 2009, 4332 RSC; (b) M. Levitus, K. Schmieder, H. Ricks, K. D. Shimizu, U. H. F. Bunz and M. A. Garcia-Garibay, J. Am. Chem. Soc., 2001, 123, 4259 CrossRef CAS PubMed.
- (a) S. P. Anthony, S. Varughese and S. M. Draper, J. Phys. Org. Chem., 2010, 23, 1074 CrossRef CAS PubMed; (b) S. P. Anthony, S. Varughese and S. M. Draper, Chem. Commun., 2009, 7500 RSC.
- S. Varughese, J. Mater. Chem. C, 2014, 2, 3499 RSC.
- T. Kawasaki, T. Kamata, H. Ushijima, S. Murata, F. Mizukami, Y. Fujii and Y. Usui, Mol. Cryst. Liq. Cryst., 1996, 286, 257 CrossRef PubMed.
- (a) G. Dong, H. Cheng, D. Chun-Ying, Q. Chun-Qi and M. Qing-Jin, New J. Chem., 2002, 26, 796 RSC; (b) J. Hamblin, A. Jackson, N. W. Alcock and M. J. Hannon, J. Chem. Soc., Dalton Trans., 2002, 1635 RSC; (c) G. Dong, P. Ke-liang, D. Chun-ying, H. Cheng and M. Qing-jin, Inorg. Chem., 2002, 41, 5978 CrossRef CAS PubMed; (d) G. Dong, P. Ke-Liang, D. Chun-Ying, Z. Yong-Gang and M. Qing-Jin, Chem. Lett., 2002, 1014 CrossRef CAS; (e) F. Tuna, J. Hamblin, A. Jackson, G. Clarkson, N. W. Alcock and M. J. Hannon, Dalton Trans., 2003, 2141 RSC; (f) Y.-F. Yue, E.-Q. Gao, C.-J. Fang, Z. He, S.-Q. Bai and C.-H. Yan, Polyhedron, 2006, 25, 2778 CrossRef CAS PubMed; (g) T.-H. Huang, M.-H. Zhang, M.-L. Tao and X.-J. Wang, Synth. React. Inorg., Met.-Org., Nano-Met. Chem., 2014, 44, 986 CrossRef CAS PubMed; (h) E.-Q. Gao, S.-Q. Bai, Z.-M. Wang and C.-H. Yan, J. Am. Chem. Soc., 2003, 125, 4984 CrossRef CAS PubMed; (i) Y.-F. Yue, E.-Q. Gao, S.-Q. Bai, Z. He and C.-H. Yan, CrystEngComm, 2004, 6, 549 RSC; (j) E.-Q. Gao, Y.-F. Yue, S.-Q. Bai, Z. He and C.-H. Yan, J. Am. Chem. Soc., 2004, 126, 1419 CrossRef CAS PubMed; (k) G. Mahmoudi and A. Morsali, Polyhedron, 2008, 27, 1070 CrossRef CAS PubMed; (l) A. Mukherjee, A. Dutta, A. D. Jana and G. K. Patra, Inorg. Chim. Acta, 2013, 404, 131 CrossRef CAS PubMed.
- (a) H. W. Smith, Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem., 1975, 31, 2701 CrossRef; (b) F. J. Lahoz, A. Tiripicchio, F. Neve and M. Ghedini, J. Chem. Soc., Dalton Trans., 1989, 1075 RSC; (c) J. Granifo, M. E. Vargas, E. S. Dodsworth, D. H. Farrar, S. S. Fielder and A. B. P. Lever, J. Chem. Soc., Dalton Trans., 1996, 4369 RSC; (d) D. A. Edwards, G. M. Hoskins, M. F. Mahon, K. C. Molloy and G. R. G. Rudolph, Polyhedron, 1998, 17, 2321 CrossRef CAS; (e) D. A. Edwards, G. M. Hoskins, M. F. Mahon, K. C. Molloy and G. R. G. Rudolph, Polyhedron, 1998, 17, 2321 CrossRef CAS; (f) E. C. Kesslen, W. B. Euler and B. M. Foxman, Chem. Mater., 1999, 11, 336 CrossRef CAS; (g) G. Dong, D. Chun-ying, F. Chen-jie and M. Qing-jin, J. Chem. Soc., Dalton Trans., 2002, 834 RSC; (h) G. Dong, H. Cheng, D. Chun-Ying, Q. Chun-Qi and M. Qing-Jin, New J. Chem., 2002, 26, 796 RSC; (i) J. Hamblin, A. Jackson, N. W. Alcock and M. J. Hannon, J. Chem. Soc., Dalton Trans., 2002, 1635 RSC; (j) M. Chandra, A. N. Sahay, S. M. Mobin and D. S. Pandey, J. Organomet. Chem., 2002, 658, 43 CrossRef CAS; (k) F. Tuna, J. Hamblin, A. Jackson, G. Clarkson, N. W. Alcock and M. J. Hannon, Dalton Trans., 2003, 2141 RSC; (l) N. M. Shavaleev, Z. R. Bell, G. Accorsi and M. D. Ward, Inorg. Chim. Acta, 2003, 351, 159 CrossRef CAS; (m) A. Singh, M. Chandra, A. N. Sahay, D. S. Pandey, K. K. Pandey, S. M. Mobin, M. C. Puerta and P. Valerga, J. Organomet. Chem., 2004, 689, 1821 CrossRef CAS PubMed; (n) E.-Q. Gao, Y.-F. Yue, S.-Q. Bai, Z. He and C.-H. Yan, J. Am. Chem. Soc., 2004, 126, 1419 CrossRef CAS PubMed; (o) Y.-F. Yue, E.-Q. Gao, S.-Q. Bai, Z. He and C.-H. Yan, CrystEngComm, 2004, 6, 549 RSC; (p) S. K. Singh, M. Chandra, D. S. Pandey, M. C. Puerta and P. Valerga, J. Organomet. Chem., 2004, 689, 3612 CrossRef CAS PubMed; (q) M.-M. Yu, W.-F. Fu, Z.-X. Li, Z.-F. Yao and L.-F. Jia, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2004, 60, m1897 CAS; (r) K. S. Singh, Y. A. Mozharivskyj, C. Thone and M. R. Kollipara, J. Organomet. Chem., 2005, 690, 3720 CrossRef CAS PubMed.
- (a) A. R. Kennedy, K. G. Brown, D. Graham, J. B. Kirkhouse, M. Kittner, C. Major, C. J. McHugh, P. Murdoch and W. E. Smith, New J. Chem., 2005, 29, 826 RSC; (b) X.-H. Zhou, T. Wu and D. Li, Inorg. Chim. Acta, 2006, 359, 1442 CrossRef CAS PubMed; (c) G. A. Broker and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2007, 63, m2420 CAS; (d) R. Karmakar, C. R. Choudhury, S. R. Batten and S. Mitra, J. Mol. Struct., 2007, 826, 75 CrossRef CAS PubMed; (e) G. Mahmoudi, A. Morsali, A. D. Hunter and M. Zeller, CrystEngComm, 2007, 9, 704 RSC; (f) G. A. Broker, R. P. A. Bettens and E. R. T. Tiekink, CrystEngComm, 2008, 10, 879 RSC; (g) P. Poplaukhin and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2008, 64, 1176 Search PubMed; (h) G. Mahmoudi and A. Morsali, Cryst. Growth Des., 2008, 8, 391 CrossRef CAS; (i) G. Mahmoudi and A. Morsali, Polyhedron, 2008, 27, 1070 CrossRef CAS PubMed; (j) S. D. Dwivedi, A. K. Singh, S. K. Singh, S. Sharma, M. Chandra and D. Pandey, Eur. J. Inorg. Chem., 2008, 5666 CrossRef CAS PubMed; (k) Z.-F. Yao, X. Gan and W.-F. Fu, Cent. Eur. J. Chem., 2008, 6, 613 CrossRef CAS PubMed; (l) S.-M. Chi, Y.-F. Wang, X. Gan, D.-H. Wang and W.-F. Fu, Cent. Eur. J. Chem., 2009, 7, 923 CrossRef CAS; (m) L. Yang, Y. Xie, J. Zou, J. Jia and X. Hong, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2009, 65, m1017 CAS; (n) X. Gan, Z.-F. Yao, J.-F. Zhang, Z. Li and W.-F. Fu, J. Coord. Chem., 2010, 63, 2800 CrossRef CAS PubMed; (o) M. Suresh, A. K. Mandal, S. Saha, E. Suresh, A. Mandoli, R. D. Liddo, P. P. Parnigotto and A. Das, Org. Lett., 2010, 12, 5406 CrossRef CAS PubMed; (p) H. D. Arman, T. Kaulgud and E. R. T. Tiekink, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, o2356 CAS; (q) E. C. Constable, G. Zhang, C. E. Housecroft and J. A. Zampese, CrystEngComm, 2010, 12, 3724 RSC.
- (a) S. Gourbatsis, J. C. Plakatouras, V. Nastopoulos, C. J. Cardin and N. Hadjiliadis, Inorg. Chem. Commun., 1999, 2, 468 CrossRef CAS; (b) M. Louloudi, V. Nastopoulos, S. Gourbatsis, S. P. Perlepes and N. Hadjiliadis, Inorg. Chem. Commun., 1999, 2, 479 CrossRef CAS; (c) S. Banerjee, J. Gangopadhyay, C.-Z. Lu, J.-T. Chen and A. Ghosh, Eur. J. Inorg. Chem., 2004, 2533 CrossRef CAS PubMed; (d) S. Banerjee, A. Ghosh, B. Wu, P.-G. Lassahn and C. Janiak, Polyhedron, 2005, 24, 593 CrossRef CAS PubMed; (e) A. D. Khalaji, M. Amirnasr and R. Welter, Anal. Sci.:X-Ray Struct. Anal. Online, 2006, 22, x151 CrossRef CAS; (f) A. D. Khalajii, M. Amirnasr and R. Welter, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, m2950 CAS; (g) A. Mentes, S. Sezek, M. E. Hanhan and O. Buyukgungor, Turk. J. Chem., 2007, 31, 667 CAS; (h) A. D. Khalaji, M. Amirnasr and R. Welter, Koord. Khim., 2010, 36, 846 Search PubMed; (i) S. Mandal, T. K. Karmakar, A. Ghosh, M. Fleck and D. Bandyopadhyay, Polyhedron, 2011, 30, 790 CrossRef CAS PubMed; (j) L. Hashemi and A. Morsali, Inorg. Chim. Acta, 2012, 386, 56 CrossRef CAS PubMed.
- (a) G. D. Smith, C. N. Caughlan, M. -Ul-Haque and F. A. Hart, Inorg. Chem., 1973, 12, 2654 CrossRef CAS; (b) A. M. Arif, F. A. Hart, M. B. Hursthouse, M. Thornton-Pett and W. Zhu, J. Chem. Soc., Dalton Trans., 1984, 2449 RSC; (c) P. K. Bowyer, K. A. Porter, A. D. Rae, A. C. Willis and S. B. Wild, Chem. Commun., 1998, 1153 RSC; (d) P. K. Pal, S. Chowdhury, P. Purkayastha, D. A. Tocher and D. Datta, Inorg. Chem. Commun., 2000, 3, 585 CrossRef CAS; (e) L. Xu, J. Perreroy, B. O. Patrick and C. Orvig, Inorg. Chem., 2001, 40, 2005 CrossRef CAS PubMed; (f) M. G. B. Drew, M. R. S. J. Foreman, M. J. Hudson and K. F. Kennedy, Inorg. Chim. Acta, 2004, 357, 4102 CrossRef CAS PubMed; (g) S. H. Rahaman, R. Ghosh, G. Mostafa and B. K. Ghosh, Inorg. Chem. Commun., 2005, 8, 1137 CrossRef CAS PubMed; (h) X.-H. Zhou, T. Wu and D. Li, Inorg. Chim. Acta, 2006, 359, 1442 CrossRef CAS PubMed; (i) N. C. Habermehl, P. M. Angus, N. L. Kilah, L. Noren, A. D. Rae, A. C. Willis and S. B. Wild, Inorg. Chem., 2006, 45, 1445 CrossRef CAS PubMed; (j) T. K. Karmakar, G. Aromi, B. K. Ghosh, A. Usman, H.-K. Fun, T. Mallah, U. Behrens, X. Solans and S. K. Chandra, J. Mater. Chem., 2006, 16, 278 RSC; (k) S. H. Rahaman, R. Ghosh and B. K. Ghosh, Inorg. Chem. Commun., 2006, 9, 1011 CrossRef CAS PubMed; (l) M. Habib, T. K. Karmakar, G. Aromi, J. Ribas-Arino, H.-K. Fun, S. Chantrapromma and S. K. Chandra, Inorg. Chem., 2008, 47, 4109 CrossRef CAS PubMed; (m) I. I. Ebralidze, G. Leitus, L. J. W. Shimon, Y. Wang, S. Shaik and R. Neumann, Inorg. Chim. Acta, 2009, 362, 4713 CrossRef CAS PubMed; (n) K. Ha, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2010, 66, m255 CAS; (o) L. Hashemi and A. Morsali, J. Coord. Chem., 2011, 64, 4088 CrossRef CAS PubMed; (p) S.-Q. Bai, C.-J. Fang, Z. He, E.-Q. Gao, C.-H. Yan and T. S. A. Hor, Dalton Trans., 2012, 13379 RSC; (q) S. Roy, S. Choubey, K. Bhar, S. Khan, P. Mitra and B. K. Ghosh, J. Mol. Struct., 2013, 1051, 328 CrossRef CAS PubMed.
- (a) A. K. Boudalis, J.-M. Clemente-Juan, F. Dahan and J.-P. Tuchagues, Inorg. Chem., 2004, 43, 1574 CrossRef CAS PubMed; (b) B. Samanta, J. Chakraborty, R. K. B. Singh, M. K. Saha, S. R. Batten, P. Jensen, M. S. E. Fallah and S. Mitra, Polyhedron, 2007, 26, 4354 CrossRef CAS PubMed.
- (a) J. Lange, H. Elias, H. Paulus, J. Muller and U. Weser, Inorg. Chem., 2000, 39, 3342 CrossRef CAS; (b) C. R. Choudhury, S. K. Dey, S. Mitra, N. Mondal, J. Ribas and K. M. A. Malik, Bull. Chem. Soc. Jpn., 2004, 77, 959 CrossRef CAS; (c) S. H. Rahaman, H.-K. Fun and B. K. Ghosh, Polyhedron, 2005, 24, 3091 CrossRef CAS PubMed; (d) H. Chowdhury, S. H. Rahaman, R. Ghosh, T.-H. Lu and B. K. Ghosh, Indian J. Chem., Sect. A: Inorg., Bio-inorg., Phys., Theor. Anal. Chem., 2005, 44, 1565 Search PubMed; (e) T. K. Karmakar, G. Aromi, B. K. Ghosh, A. Usman, H.-K. Fun, T. Mallah, U. Behrens, X. Solans and S. K. Chandra, J. Mater. Chem., 2006, 16, 278 RSC; (f) T. K. Karmakar, M. Ghosh, M. Fleck, G. Pilet and D. Bandyopadhyay, J. Coord. Chem., 2012, 65, 2612 CrossRef CAS PubMed.
- Oxford Diffraction, CrysAlis PRO, Oxford Diffraction Ltd., Yarnton, Oxfordshire, England, 2010 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
- W. Tang, Y. Xiang and A. Tong, J. Org. Chem., 2009, 74, 2163 CrossRef CAS PubMed.
- J. B. Birks, Photophysics of Aromatic Molecules, Wiley, London, 1970 Search PubMed.
- S. R. LaPlante, R. Carson, J. Gillard, N. Aubry, R. Coulombe, S. Bordeleau, P. Bonneau, M. Little, J. O'Meara and P. L. Beaulieu, J. Med. Chem., 2013, 56, 5142 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Crystallographic data; IR, 1H NMR, 13C NMR, NOESY, UV absorption spectra, excitation spectra, photo luminescence spectra and other supplementary material. CCDC 963345, 962823 and 930055. For ESI and crystallographic data in CIF or other electronic format See DOI: 10.1039/c5ra09562j |
| This journal is © The Royal Society of Chemistry 2015 |