
Self-assembled capsules of poly-N-glycidyl histidine ether–tannic acid for inhibition of biofilm formation in urinary catheters†
Denial Mahata,
Santi M. Mandal,
Amit Basak and
Golok B. Nando*
Department of Chemistry, Rubber Technology Centre, Central Research Facility, Indian Institute of Technology Kharagpur, Kharagpur 721302, India. E-mail: golokb@rtc.iitkgp.ernet.in; Fax: +91 3222 282252; Tel: +91 3222 283194, +91 3222 283300
First published on 28th July 2015
Abstract
A hydrophilic biocompatible polymer, N-glycidyl histidine ether, was synthesized and self-assembled after complexation with tannic acid to form unique toroidal-like capsules, which when coated on a urinary catheter significantly reduce the association of urinary pathogens and inhibit biofilm formation. Thus, the coated catheter is effective for long-term use and hinders catheter-associated infections.
Introduction
The self-assembly of polymeric compounds have been extensively explored as well-defined structures for their potential biomedical applications in regenerative medicine, gene delivery, coating on medical devices, tissue engineering and for the controlled release of drugs.1–3 The self-assembled structures with their tunable activity or novel properties depend upon the nature of the polymeric segments4 or hybrid materials used.5 Several polymeric materials have been well characterized for their defined structures and dynamics.6 The designing of polymeric materials is crucial for the next generation of targeted and biocompatible nanostructures to bridge the gap between materials and life sciences. In general, defined polymeric nanostructures are formed using suitable amphiphilic polymers by varying the molecular weight and chemical structure and through self-assembly in an aqueous environment via hydrophobic or non-covalent interactions.7 There is a limit on polymer-based nanostructures in clinical applications due to their non-specific toxicity. These factors need to be taken into account, and thus we synthesized a histidine-based polymer for possible clinical application as a coating material on medical devices. Histidine-based copolymers have recently been used in gene and drug delivery due to their non-toxic nature.8The urinary catheter is one of the most commonly used medical devices in urology. Catheterization facilitates the drainage of patient's urine from the bladder and can be used as an indwelling or in an intermittent manner according to the patient's requirement. An indwelling catheter is the major cause of catheter-associated urinary tract nosocomial infections.9 Polymers such as polyvinyl chloride, polyethylene, polyurethane and silicone are mostly used in the production of catheters.10–12 Several approaches have been undertaken to modify the outer surface of the catheter by employing silver nanoparticles, a silver-based alloy or different types of antibacterial hydrogel to prevent biofilm formation by urinary tract infection (UTI) pathogens.13–16 Silver-based materials are used for the coating because silver has been known as an antibacterial agent for centuries and used in major applications in wound care management.17 The safety of silver ion for internal use is still unclear. Prolonged urinary tract infections or long-term catheter use facilitate the formation of catheter biofilms. Catheter biofilms constitute complex microbial communities that are difficult to remove or to prevent their association and result in uncontrolled bacteriuria.18,19 Herein, we developed a biocompatible N-glycidyl histidine ether polymer hybridized with tannic acid-based self-assembled capsular structures for coating catheters to prevent biofilm formation by UTI-associated pathogens.
L-Histidine hydrochloride monohydrate (1.05 g, 0.005 molar) was dissolved in 150 mL ethanol at pH 8.0 by the addition of NaOH (0.006 molar) at 40 °C for 5 hours and then epichlorohydrin (2 mL) was added dropwise. The pH of the solution was further increased by the addition of NaOH and the reaction mixture was refluxed for 24 hours at 55 °C. A solid reddish resinous product was obtained and isolated by filtration. The solid product was dissolved in water and neutralized by dilute HCl, and then precipitated in methanol (Scheme 1). The polymer was characterized using UV-vis, FTIR and 1H NMR (Fig. S1 and S2†). The UV-visible spectrum of the polymer shows a π–π* transition at 212 nm and a weak, broad band for a n–π* transition at 292 nm. FT-IR analysis of pure L-histidine shows a sharp absorption at 3410 cm−1 with different submaxima at 3016, 2625 and 2300 cm−1 for the NH group of the imidazole ring because of the Fermi resonance of the vNH fundamental with various combinations and overtones of the internal vibrations. The peaks at 3277 and 3142 cm−1 correspond to the antisymmetric and symmetric stretching mode for the primary NH2 group in L-histidine.20 In addition, distinguishable bands for imidazole NH and free NH2 are observed at 876 cm−1 and 832 cm−1 due to the vibration and wagging of vibration of the amine group, respectively. However, the disappearance of the submaxima of the imidazole –NH group signal and the appearance of only the symmetric stretching frequency of the primary NH2 group in the polymer strongly suggest that epichlorohydrin has reacted with the imidazolium ring –NH in the histidine molecule. Moreover, the appearance of a new peak at 1082 cm−1 is due to the presence of C–O–C symmetric stretch vibrations. In the 1H NMR spectrum, a doublet peak at 3.2 ppm is related to –CH2– and one at 3.92 ppm corresponds to the –CH![[double bond splayed right]](https://www.rsc.org/images/entities/char_e00a.gif) proton. Singlet signals were observed at 7.39 and 8.6 ppm for CH–N (a) and CH–N (b), respectively, in the imidazole ring. The signals at 4.4–4.8 ppm (m, 2H) were due to CH2 attached to the imidazole moiety, whereas the signal at 3.2 ppm (m, 2H) was due to the –O–CH2–CH– unit. The multiplet at 3.6 ppm was attributed to the
proton. Singlet signals were observed at 7.39 and 8.6 ppm for CH–N (a) and CH–N (b), respectively, in the imidazole ring. The signals at 4.4–4.8 ppm (m, 2H) were due to CH2 attached to the imidazole moiety, whereas the signal at 3.2 ppm (m, 2H) was due to the –O–CH2–CH– unit. The multiplet at 3.6 ppm was attributed to the ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) CH– group. The polymer was characterized with MALDI-TOF-MS analysis. Spectra obtained from the GHEP polymer showed a series of macromolecular masses as m/z 696, m/z 656, 1079, 1289, and 1478. A mass distribution analysis of the polymer evidenced a mixture of both the linear and cyclic forms of GHEP. The accurate mass of m/z 696 represents the linear form of GHEP and showed a prominent fragment at m/z 292, corresponding to the N-glycidyl histidine ether monomeric unit with a sodium ion (Fig. 1). The masses of m/z 656, 1079, and 1289 are comparable to the cyclic form of GHEP, which looks like the crown ether compounds and that forms a complex with the sodium ion. Their corresponding chemical structures are described in the ESI (Fig. S3†). The TGA and DTG curves of L-histidine and GHEP are shown in Fig. S4.† L-Histidine hydrochloride monohydrate shows degradation first from 180 °C to 200 °C due to the loss of water molecules and a considerable thermal stability up to 280 °C, whereas above 360 °C, most of the sample is totally degraded on heating.21 However, a two-step degradation is seen in the DTG curve of GHEP, in which the first-stage degradation is loss of water molecules and second is degradation at 330 °C, where Tmax is obtained at 366 °C. The tailing effect of the sample is prominent above 415 °C. Therefore, the degradation of GHEP begins at a much higher temperature than for histidine. This behavior could originate from the presence of a poly-ether linkage in the polymer.22 The GHEP and tannic acid (TA) aqueous solutions are mixed in different ratios and allowed for self-assembly. At a 1
CH– group. The polymer was characterized with MALDI-TOF-MS analysis. Spectra obtained from the GHEP polymer showed a series of macromolecular masses as m/z 696, m/z 656, 1079, 1289, and 1478. A mass distribution analysis of the polymer evidenced a mixture of both the linear and cyclic forms of GHEP. The accurate mass of m/z 696 represents the linear form of GHEP and showed a prominent fragment at m/z 292, corresponding to the N-glycidyl histidine ether monomeric unit with a sodium ion (Fig. 1). The masses of m/z 656, 1079, and 1289 are comparable to the cyclic form of GHEP, which looks like the crown ether compounds and that forms a complex with the sodium ion. Their corresponding chemical structures are described in the ESI (Fig. S3†). The TGA and DTG curves of L-histidine and GHEP are shown in Fig. S4.† L-Histidine hydrochloride monohydrate shows degradation first from 180 °C to 200 °C due to the loss of water molecules and a considerable thermal stability up to 280 °C, whereas above 360 °C, most of the sample is totally degraded on heating.21 However, a two-step degradation is seen in the DTG curve of GHEP, in which the first-stage degradation is loss of water molecules and second is degradation at 330 °C, where Tmax is obtained at 366 °C. The tailing effect of the sample is prominent above 415 °C. Therefore, the degradation of GHEP begins at a much higher temperature than for histidine. This behavior could originate from the presence of a poly-ether linkage in the polymer.22 The GHEP and tannic acid (TA) aqueous solutions are mixed in different ratios and allowed for self-assembly. At a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4 ratio (TA:GHEP), yellowish GHEP turns into white colloidal dispersions due to their self-assembly mechanism (Fig. 2a). Spectral characterization confirmed the formation of hydrogen-bonding between the polymer and tannic acid in the complex in an aqueous solution at pH 7.0 (Fig. S5 and S6†). The spectral analysis of GHEP shows a sharp π–π* transition at 212 nm and a weak, broad band for a n–π* transition at 292 nm. It has been reported that pure tannic acid displays two characteristic absorption peaks at 279 nm due to a n–π* transition of the phenolic –OH group and a π–π* transition at 212 nm in the presence of an aromatic chromophore. However, the sharp n–π* transition band of tannic acid is broadened and red-shifted from 274 to 283 nm in the tannic acid–GHEP self-assembled capsule. This may be due to the presence of a hydrogen-bonding interaction between tannic acid and GHEP. The π–π* transition also shows a red-shift from 212 nm to 214 nm, which can be attributed as a π–π interaction between benzene and the imidazole ring. Therefore, λmax is shifted towards a higher wavelength with the successive addition of polymer concentration in tannic acid solution (Fig. S7†).
:4 ratio (TA:GHEP), yellowish GHEP turns into white colloidal dispersions due to their self-assembly mechanism (Fig. 2a). Spectral characterization confirmed the formation of hydrogen-bonding between the polymer and tannic acid in the complex in an aqueous solution at pH 7.0 (Fig. S5 and S6†). The spectral analysis of GHEP shows a sharp π–π* transition at 212 nm and a weak, broad band for a n–π* transition at 292 nm. It has been reported that pure tannic acid displays two characteristic absorption peaks at 279 nm due to a n–π* transition of the phenolic –OH group and a π–π* transition at 212 nm in the presence of an aromatic chromophore. However, the sharp n–π* transition band of tannic acid is broadened and red-shifted from 274 to 283 nm in the tannic acid–GHEP self-assembled capsule. This may be due to the presence of a hydrogen-bonding interaction between tannic acid and GHEP. The π–π* transition also shows a red-shift from 212 nm to 214 nm, which can be attributed as a π–π interaction between benzene and the imidazole ring. Therefore, λmax is shifted towards a higher wavelength with the successive addition of polymer concentration in tannic acid solution (Fig. S7†).
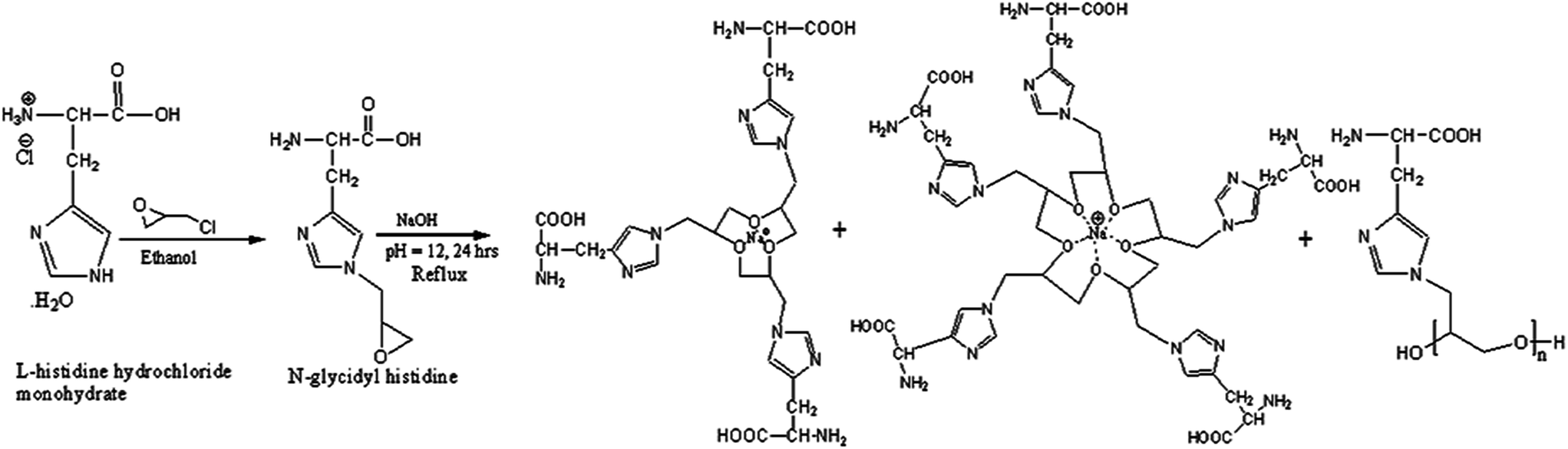 | ||
| Scheme 1 Synthesis route of N-glycidyl histidine ether polymer. | ||
 | ||
| Fig. 1 MALDI TOF-MS analysis of the synthesized polymer representing their cyclic structures. | ||
 | ||
| Fig. 2 Microscopic characterization of a N-glycidyl histidine ether polymer self-assembled with tannic acid. Macroscopic solution visual images of the synthesized polymer GHEP (a, left glass vial) and GHEP self-assembled with tannic acid (a, right glass vial). Particle size distribution obtained from DLS analysis (b). FE-SEM image obtained from GHEP self-assembled with tannic acid (c) and their enlarged view (d). | ||
The FTIR spectrum of pure tannic acid shows a broad signal at 3356 cm−1 along with sharp band at 1200 cm−1 due to phenolic –OH stretching and the deformation mode of vibration. The bands at 1730 cm−1 and 1322 cm−1 are attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–O stretching frequencies in the ester group. The spectrum of the GHEP polymer and self-assembled GHEP/tannic acid also shows different band shifting, which is an evidence of the hydrogen-bonding interaction in the capsule. The shifting of the –OH band stretching from 3412 to 3401 cm−1 and the asymmetric –NH stretching frequency from 3142 to 3132 cm−1 at lower frequency suggests their intermolecular hydrogen-bonding interaction. The CO and C–O stretching of carboxylic acid at 1724 cm−1 and 1389 cm−1 are also shifted to 1709 cm−1 and 1354 cm−1, respectively. Moreover, the characteristic C–O–C symmetric stretch peak shift from 1082 cm−1 to 1042 cm−1 can be attributed to the molecular association through hydrogen-bonding in the ether unit.
O and C–O stretching frequencies in the ester group. The spectrum of the GHEP polymer and self-assembled GHEP/tannic acid also shows different band shifting, which is an evidence of the hydrogen-bonding interaction in the capsule. The shifting of the –OH band stretching from 3412 to 3401 cm−1 and the asymmetric –NH stretching frequency from 3142 to 3132 cm−1 at lower frequency suggests their intermolecular hydrogen-bonding interaction. The CO and C–O stretching of carboxylic acid at 1724 cm−1 and 1389 cm−1 are also shifted to 1709 cm−1 and 1354 cm−1, respectively. Moreover, the characteristic C–O–C symmetric stretch peak shift from 1082 cm−1 to 1042 cm−1 can be attributed to the molecular association through hydrogen-bonding in the ether unit.
FE-SEM analysis of the colloidal particle reveals the toroidal capsule-like structure with a smooth surface (Fig. 2c and d). The TEM image shows that the capsules are porous in nature (Fig. S8†). Dynamic light scattering (DLS) analysis of the self-assembled capsules revealed that the average particle size distribution covered a broad range from 100 to 550 nm (Fig. 2b). The average surface zeta-potential values of tannic acid and GHEP polymer in an aqueous solution were +10.8 mV and +2.31 mV, respectively (Fig. S9†). However, the average surface zeta-potential value +33.2 mV, of the capsule in a colloidal solution suggests that the self-assembled particle is stable, which is also confirmed by FE-SEM analysis.
The GHEP–TA/PVP composite coated on the outer surface of the catheter was characterized with FE-SEM and AFM analysis. Fig. 3 shows the surface topography obtained from both the coated (Fig. 3a and c) and uncoated (Fig. 3b and d) catheter. The uncoated surface shows a comparatively rough surface morphology with the defined structure of the natural rubber latex catheter. After coating with the composite, the surface was smoother with a heterogeneous distribution of the capsules over the catheter surface. The appearances of such morphological changes are indicative of composite coating. Furthermore, a release study was conducted to determine the release of composite in a natural urine environment. It was observed that a maximum of 24% of the composite was released within 15 days incubation in urine (Fig. S10†). The slow release might be due to the swelling of PVP in the aqueous phase.23 The degradation of the composite was also studied through TGA analysis (Fig. S11†). The TGA analysis of the composite showed two stages of degradation, wherein the total weight loss in the first stage is enhanced from 10% to 21%. The maximum degradation temperature (Tmax) shifted towards a higher value from 220 °C to 230 °C after 15 days of incubation in urine. The Tmax value of the control coated-material observed at 220 °C corresponded to the degradation of tannic acid.24 The increased rate of degradation and enhanced Tmax value suggests that tannic acid degrades into a strong multifunctional crosslinker25 cellulose and gallic acid with increasing incubation time. The second degradation stage showed no prominent shift of Tmax at 449 °C associated to PVP.
 | ||
| Fig. 3 FE-SEM and AFM images obtained from a latex catheter. FE-SEM image of the outer surface of a catheter without a coating (a) and coated with the self-assembled capsule/PVP composite (b). AFM image of the outer surface of a catheter without coating (c) and coated with the self-assembled capsule/PVP composite (d). | ||
Both the coated and uncoated catheter were subjected to biofilm formation by a uropathogen, E. coli strain no. 21.26 After 48 h of incubation, the catheters were removed from the culture, gently washed with PBS (1×), and visualized under FE-SEM. Fig. 4a shows the strong adherence of bacterial biofilm over the surface of the uncoated catheter, whereas bacterial adherence was significantly less and almost absent on the coated catheter (Fig. 4b). A viable colony count assay was also performed from the detached biofilm of both (coated and uncoated) catheter surfaces, and less attachment of bacterial cells to the coated catheter surface was observed in comparison to the uncoated surface (Fig. 4d). The initial catheter surface (natural rubber latex) was considerably hydrophobic and showed a water contact angle of 86.2° ± 2.5°. After treatment of the surface with GHEP–TA/PVP composite, water contact angle on the outer surface of the catheter was 53.9° ± 1.2°, which is in good agreement with the contact angle of the GHEP–TA/PVP composite (56.4° ± 1.5°) (Fig. 4c).
 | ||
| Fig. 4 FE-SEM image obtained from a latex catheter after bacterial biofilm formation and characterization of the catheter surface. FE-SEM image of the uncoated catheter shows strong bacterial adhesion to the surface after one week grown in E. coli culture (a), whereas no biofilm was visualized from the coated catheter surface (b). Various contact angle measurements of only the self-assembled capsule/PVP composite after film formation (i), uncoated latex catheter (ii), and coated with the self-assembled capsule/PVP composite (iii). The biofilm was quantified with CV stain (d, grey colour) and by quantification of the viable colony from the removed biofilm of both the coated and uncoated catheter surface. MTT analysis (e) of the synthesized GHEP polymer (open circle) and the self-assembled capsule (closed circle) revealed that the self-assemble structure is less toxic, and IC50 values were observed at a concentration of >500 μg mL−1. | ||
Thus, the hydrophilic surface of the coated catheter can be attributed to the abundance of hydrophilic groups, especially hydroxyl from tannic acid and carbonyl groups from PVP. Therefore, it is clear that the hydrophobicity of the uncoated surface was greater than the coated surface. Earlier, Cerca et al.27 reported that adhesion to the hydrophobic substrata for bacteria occurred to a greater extent than to hydrophilic surfaces. Finally, data obtained from the MTT assay revealed that the polymer (Fig. 4e), GHEP and capsules all had a minimum toxicity at higher concentration of 500 μg mL−1 against the 3T3 non-carcinoma cell line. Therefore, the coating capsule material is biocompatible in clinical application.
In conclusion, the N-glycidyl histidine ether self-assembled with tannic acid in an aqueous phase (pH 7.0) and extended to a toroidal capsule-like microstructure. Urinary catheters coated with capsules have the potential for long-term catheterization without biofilm-mediated serious infection and encrustation.
Acknowledgements
D. Mahata acknowledges the University Grant Commission (UGC), Delhi, India for senior research fellowship. A. Basak acknowledges the Department of Science and Technology (DST) for J. C. Bose National Fellowship. We are grateful to IIT-Kharagpur for providing all the necessary facilities.Notes and references
- A. R. Hirst, B. Escuder, J. F. Miravet and D. K. Smith, Angew. Chem., Int. Ed., 2008, 47, 8002–8018 CrossRef CAS PubMed.
- A. Roy, O. L. Franco and S. M. Mandal, Curr. Protein Pept. Sci., 2013, 14, 580–587 CrossRef CAS.
- M. Yokoyama, Expert Opin. Drug Delivery., 2010, 7, 145e58 Search PubMed.
- A. Blanazs, S. P. Armes and A. J. Ryan, Macromol. Rapid Commun., 2009, 30, 267–277 CrossRef CAS PubMed.
- D. Mahata, S. M. Mandal and G. B. Nando, RSC Adv., 2014, 4, 48559–48562 RSC.
- E. Busseron, Y. Ruff, E. Moulin and N. Giuseppone, Nanoscale, 2013, 5, 7098–7140 RSC.
- T. S. Kale, A. Klaikherd, B. Popere and S. Thayumanavan, Langmuir, 2009, 25, 9660–9670 CrossRef CAS PubMed.
- H. Wu, I. Zhu and V. P. Torchilin, Biomaterials, 2013, 34, 1213–1222 CrossRef CAS PubMed.
- P. A. Tambyah, K. T. Halvorson and D. G. Marki, Mayo Clin. Proc., 1999, 74, 131–136 CrossRef CAS PubMed.
- S. Noimark, C. W. Dunnill, C. W. M. Kay, S. Perni, P. Prokopovich, S. Ismail, M. Wilsond and I. P. Parkin, J. Mater. Chem., 2012, 22, 15388 RSC.
- B. Mishra, A. Basu, R. R. Y. Chua, R. Saravanan, P. A. Tambyah, B. Ho, M. W. Change and S. S. J. Leong, J. Mater. Chem. B, 2014, 2, 1706 RSC.
- N. MacCallum, C. Howell, P. Kim, D. Sun, R. Friedlander, J. Ranisau, O. Ahanotu, J. J. Lin, A. Vena, B. Hatton, T. S. Wong and J. Aizenberg, ACS Biomater. Sci. Eng., 2015, 1, 43–51 CrossRef CAS.
- F. D. Matl, A. Obermeier, S. Repmann, W. Friess, A. Stemberger and K. D. Kuehn, Antimicrob. Agents Chemother., 2008, 52, 1957–1963 CrossRef CAS PubMed.
- L. Cormio, P. La Forgia, D. La Forgia, A. Siitonen and M. Ruutu, Eur. Urol., 2001, 40, 354–359 CrossRef CAS PubMed.
- K. Davenport and F. X. Keeley, J. Hosp. Infect., 2005, 60, 298–303 CrossRef CAS PubMed.
- K. Davis, Am. J. Infect. Control, 2005, 33, 55–56 CrossRef PubMed.
- A. Lansdown, Curr. Probl. Dermatol., 2006, 33, 17–34 CAS.
- D. Stickler, R. Young, G. Jones, N. Sabbuba and N. Morris, Urol. Res., 2003, 31, 306–311 CrossRef PubMed.
- S. Noimark, C. W. Dunnill, M. Wilson and I. P. Parkin, Chem. Soc. Rev., 2009, 38, 3435–3448 RSC.
- A. K. Rai, W. Fei, Z. Lu and Z. Lin, Theor. Chem. Acc., 2009, 124, 37–47 CrossRef CAS.
- K. D. Trimukhe and A. J. Varma, Carbohydr. Polym., 2009, 75, 63–70 CrossRef CAS PubMed.
- A. L. Brocas, C. Mantzaridis, D. Tunc and S. Carlotti, Prog. Polym. Sci., 2013, 38, 845–873 CrossRef CAS PubMed.
- D. Lubasovaab, H. Niub, X. Zhaob and T. Lin, RSC Adv., 2015, 5, 54481–54487 RSC.
- Y. Peng, Z. Zheng, P. Sun, X. Wang and T. Zhang, New J. Chem., 2013, 37, 729–734 RSC.
- S. Ma, Y. Jiang, X. Liu, L. Fan and J. Zhu, RSC Adv., 2014, 4, 23036–23042 RSC.
- T. Samanta, G. Roymahapatra, W. F. Porto, S. Seth, S. Ghorai, S. Saha, J. Sengupta, O. L. Franco, J. Dinda and S. M. Mandal, PLoS One, 2013, 8, e58346 CAS.
- N. Cerca, G. B. Pier, M. Vilanova, R. Oliveira and J. Azeredo, Res. Microbiol., 2005, 156, 506–514 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06815k |
| This journal is © The Royal Society of Chemistry 2015 |