
Efficient Au0/C catalyst synthesized by a new method for acetylene hydrochlorination†
Abstract
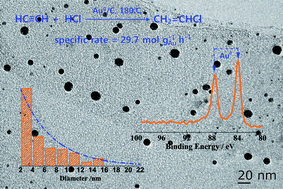
A new impregnation method, involving a mixture of solvents and a vacuum drying process, was used to prepare a gold catalyst (MIV-1Au/C1) for acetylene hydrochlorination. It was found that MIV-1Au/C1 was twice as active as the catalyst prepared through the traditional method (PI-1Au/C1). The new method is green, mild and simple. Moreover, it appears to be controllable by tuning solvent and temperature. Excellent Au dispersion in MIV-1Au/C1 was revealed by transmission electron microscopy (TEM). In addition, X-ray photoelectron spectroscopy (XPS) profiles proved that Au0 was the only active species of MIV-1Au/C1 at the initial/highest point of testing. Further XPS results showed that Au0 could be oxidized to Au3+ during the reaction along with the deactivation of MIV-1Au/C1. Thus, Au0 appeared to be preferred for the catalytic route. Our findings demonstrate that this new method has potential as a catalyst for acetylene hydrochlorination. Moreover, this study highlighted the importance of Au0 in this field.
 Please wait while we load your content...
Please wait while we load your content...

