
Fibril aggregates of the poly(glutamic acid)–drug conjugate†
Jingjing Lai and
Yanbin Huang*
Key Laboratory of Advanced Materials (MOE), Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: yanbin@tsinghua.edu.cn
First published on 27th May 2015
Abstract
Polymer–drug conjugates are an important category of nanomedicine, and the aggregate size and shape are important for the applications. The poly(glutamic acid)–doxorubicin conjugate was found to form fibrillar aggregates with diameter about 40 nm and length up to tens of microns. The fibrils slowly dissociate and release the soluble conjugates at neutral pH condition.
Introduction
Since the general concept of polymer–drug conjugates was proposed,1 they have been extensively investigated to improve the drug solubility, prolong the circulation time, and possibly target the tumor sites for small molecular anti-cancer drugs. Many such polymer–drug conjugate systems have advanced into clinic trials,2,3 among which the poly-L-glutamic acid (PGA)–paclitaxel conjugate is currently the furthest in terms of the development stage.4 PGA–paclitaxel conjugates (under various names such as CT-2103, Xyotax™, Opaxio™) had failed in one Phase III clinical trial, but the post-trial analysis showed that the conjugate treatment was actually effective in the female sub-population,5 and consequently a new Phase III clinical trial is currently going on for the treatment of ovarian cancer in women only. Compared with other polymers used for the drug conjugation, PGA is biodegradable and hence expected not to have the tissue accumulation problem.6Although there are numerous studies about their molecular design in terms of types of polymers, the linker chemistry, the polymer molecular weight and the drug loading, the aggregation behavior of polymer–drug conjugates is much less studied.7–14 In some of the studies, the polymer–drug conjugate in the aqueous solution is assumed to form unimolecular micelles,7–10 i.e., the hydrophobic drug moieties aggregate as the core with the hydrophilic polymer backbone as the corona, and hence the size is directly determined by the molecular weight of the polymer. As the polymer molecular weight usually in the order of magnitude of 104 Da, the size of these polymer–drug conjugates are about 5–25 nm in diameter.9–14 However, a larger particle size of about 50–100 nm may be optimal for long circulation and tumor accumulation by the enhanced permeability and retention (EPR) effect.15–17 For a single polymer–drug conjugate chain to reach such a large particle size, the polymer usually should have high molecular weight in the orders of 106 Da,18 which is not easy to achieve in synthesis and also difficult for the drug conjugation chemistry. This is in contrast to other drug nanoparticles (e.g. liposomes or polymer micelles), where the particles are formed via well-defined self-assembly processes and their size can be varied with relative ease.
Using a poly-L-glutamic acid–doxorubicin (PGA–Dox) conjugate as the model system (Scheme 1), here we show that controlling the aggregation state of polymer–drug conjugates seems a promising approach to decouple the particle size and the polymer molecular weight. It should be noticed that, as a model study, we used an amide linker for its chemistry simplicity. We demonstrated that the PGA–Dox conjugate can form fibril aggregates with diameter about 40 nm and length ranging from two to tens of micrometers under specific conditions.
 | ||
| Scheme 1 Molecular structure of the PGA–Dox conjugate. | ||
Experimental section
Synthesis of PGA–Dox
Doxorubicin (Dox) hydrochloride salt was purchased from Melone Pharma. Poly-L-glutamic acid (PGA) sodium salt (molecular weight 15–50 kDa) was purchased from Sigma Aldrich. 2-Ethoxy-1-ethoxycarbonyl-1,2-dihydroquinoline (EEDQ) was purchased form Alfa Aesar. N,N-dimethylformamide (DMF, super-dry) was purchased from J&K Chemical. Other reagents were purchased from Beijing Chemical Company.Dox hydrochloride salt was precipitated with 1.5 equivalents of NaHCO3 in water to obtain Dox in its base form. The dark-red precipitate was collected by centrifugation, washed with water and freeze dried. PGA sodium salt was dissolved in water, and the pH was adjusted to 2. The resulted white precipitate was collected by centrifugation, washed with water, and freeze dried to obtain PGA in its acid form.19
In a typical conjugation step,19 300 μL DMF was used to dissolve 5 mg PGA and 1.6 mg Dox in a 2 ml glass vial (glutamic acid/Dox molar ratio about 11) and stirred for about 15 min until the powder completely dissolved. Then, 0.6 mg of EEDQ was added. The reaction proceeded at room temperature for 24 hours and was monitored by TLC (methanol as the solvent; Dox, Rf = 0.8; PGA and PGA–Dox, Rf = 0). After the reaction, the mixture was poured into 1 ml chloroform and the precipitate was collected by centrifugation and dried under vacuum. The obtained powder was dissolved in 0.5 M NaHCO3 solution and the low molecular weight impurities were removed by ultracentrifugal filtration (Millipore Amicon Ultra-0.5, Ultracel-3, cut-off molecular weight 3 kDa) at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm. The remained solution was freeze dried to obtain the final product as red powder.
000 rpm. The remained solution was freeze dried to obtain the final product as red powder.
The conjugate structure was confirmed by 1H NMR (JEOL JNM-ECS400). As shown in Fig. 1, the drug contents in the conjugate were calculated by comparing the integral of C–CH3 from the Dox moiety and α-H of PGA, and it can be varied from 10–30% by changing the PGA/Dox feed ratio. Typical GPC result and diffusion ordered spectroscopy (DOSY) was shown in Fig. S1 and S2.†
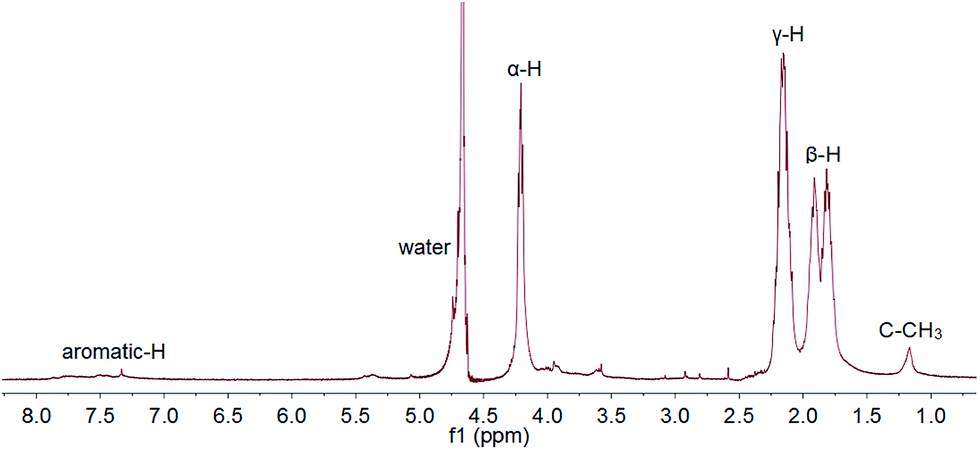 | ||
| Fig. 1 1H NMR (400 MHz, D2O) spectrum of PGA–Dox. δ = 4.35–4.05 (α-H), 2.30–2.00 (γ-H) and 2.00–1.65 ppm (β-H) are assigned to PGA. δ = 7.85–6.95 ppm (aromatic-H) and 1.20 ppm (H3C–C) are assigned to Dox. | ||
Transmission electron microscope
Transmission electron microscope (TEM) pictures were obtained with a Hitachi H-7650B (Hitachi, Japan) transmission electron microscope at an accelerating voltage of 120 kV. The sample solution was dropped on the carbon-coated copper grids and air-dried.Fluorescence spectroscopy
Fluorescence spectra were collected on a Hitachi F-7000 fluorescence spectrometer with excitation at 350 nm. The scan speed was set at 1200 nm min−1, the slit width for both excitation and emission was 5.0 nm and the PMT voltage was 400 V.Circular dichroism
Circular dichroism (CD) spectra were collected on a Pistar π-180 instrument at room temperature in a cuvette with a 0.1 cm path length. The spectra were recorded at a 1 nm interval from 280 nm to 190 nm.Results and discussion
The PGA–Dox was prepared using the EEDQ method, and the drug content in the polymer–drug conjugates could be adjusted from 10–30 wt% (i.e., about 3–12 mol% of the glutamic acid units, determined by 1H NMR). The products dissolved well in deionized water with concentration up to 20 mg ml−1 and resulted in dark-red solutions.In a preliminary test, we changed the pH of 1 mg ml−1 PGA–Dox (drug content ∼10 wt%) solution, and found the solution remained clear from pH 8.5 to pH 4.5, but turned turbid below pH 4.2. This is understandable as the polymer–drug conjugate is basically a polyanion with randomly-positioned hydrophobic side groups along the polymer chain. Then, we repeated the experiment but simultaneously monitor the fluorescence emission at 562 nm and 700 nm, respectively (Fig. 2). The fluorescence emission at 562 nm came from the Dox moieties, and the one at 700 nm basically originated from the light scattered by the aggregates formed in the solution. As shown in Fig. 2, the fluorescence intensity of the Dox moieties decreased continuously as the solution pH decreased from 8.5 to 3.4, which should be resulted from the self-quenching of the aggregating Dox moieties (Fig. S3†).20,21 In contrast, the increase of the scattering intensity at 700 nm only became significant at pH below 4.5. These results suggested the possible existence of a suitable pH range, within which the anionic charge density along the PGA chain should be low enough to allow the Dox moieties to aggregate, but at the same time not too low that large precipitate would form uncontrollably. We hypothesized that within this suitable pH range, regular aggregates of PGA–Dox might form in the aqueous solution. Subsequently, screening experiments were conducted on the solution pHs, and as shown below, this suitable pH range was found to be quite narrow, between 4.5 and 4.7.
 | ||
| Fig. 2 Fluorescence intensity of the Dox moieties (circles, left axis) and the scattering intensity from the aggregates (triangles, right axis) of 1 mg ml−1 PGA–Dox aqueous solution, excitation: 350 nm. The grey zone represents the pH range to obtain regular aggregates. | ||
As a typical example, 2 mg ml−1 PGA–Dox (drug content ∼20 wt% or 7 mol% conjugation of the glutamic acid units, the same for the following discussions unless otherwise specified) solution was incubated at pH 4.7, and the temperature was increased to 60 °C to accelerate the process. After 24 hours, red precipitates were collected by centrifugation and observed with TEM (Fig. 3). Different from the unstructured aggregates formed below pH 4.2 (data not shown), these aggregates were mostly fibrils with diameters of about 40 nm and length up to tens of microns (Fig. S4†). Samples with other drug content ratio and concentration also formed similar fibrils (Fig. S5†). A small amount of the fibrils seemed larger in diameter than others and their surfaces seemed not as smooth either. It might be due to secondary aggregates or the aggregates of detached Dox adsorbed on the primary fibril surface.
 | ||
| Fig. 3 TEM images of the collected precipitates from 2 mg ml−1 PGA–Dox solution, incubated at pH 4.7, 60 °C for 24 hours. The scale bars are 2 μm in the left picture and 500 nm in the right picture, respectively. | ||
The yield of the fibrillar aggregates kept growing as the incubation proceeded. We monitored the change of the concentration of Dox moieties in the supernatant by UV-vis spectroscopy, and used it to calculate the yield of the precipitates. As shown in Fig. 4, the amount of the aggregates increased almost linearly with time, and after two weeks, about 40 percent of the Dox moieties were found in the fibril aggregates.
 | ||
| Fig. 4 The yield of the fibrillar precipitates as a function of incubation time, detected using UV-vis spectroscopy (485 nm). | ||
These fibril aggregates are clearly different from those observed in the literature. For comparison, previous studies about the solution behavior of polymer–drug conjugates usually reported structure models of unimolecular or multimolecular micelles with diameter of ∼5–25 nm. For example, using light scattering and GPC, Ulbrich et al.7 studied the poly(N-2-hydroxypropyl-methacrylamide) (HPMA) based conjugates and suggested that micelles of ∼5 nm formed, which consisted of 1–5 polymers depending on the drug contents. Also in the HPMA system, Paul et al.10 used small-angle neutron scattering and showed that the particle size was ∼10 nm without evidences of multi-molecular aggregation, while Filippov et al.11,12 proposed a core–shell structure with diameter of 5–10 nm, and the aggregation number increased with the increasing content of the hydrophobic moieties.
After demonstrating that PGA–Dox could form fibril aggregates in the pH range of 4.5–4.7, we were concerned about the aggregate stability in the physiologically relevant pHs, i.e., whether the fibrils would dissolve too quickly to lose its advantages as aggregates in neutral pH condition. When the aggregates were re-suspended in deionized water at room temperature and the pH was adjusted to 7, the concentration of Dox in the supernatant solution grew gradually (Fig. 5, left), but the initial suspension did not become apparently clear until one week later. As shown in the TEM image in Fig. 5, most of the long fibrils disappeared after one week, but certain amount of short fibrils remained in the solution. The relatively slow dissociation process at pH 7 suggested that the fibrils were mainly stabilized by the hydrophobic association among the Dox moieties, which does not change much as pH changed. It should be noted that the dissociation process were even slower in pH 7.4 PBS solution (sodium chloride 137 mM, phosphate buffer 10 mM, potassium chloride 2 mM, data not shown), probably because the salts screened the ionic repulsion among PGA acid groups, which is supposed to be the driving force for dissociation.
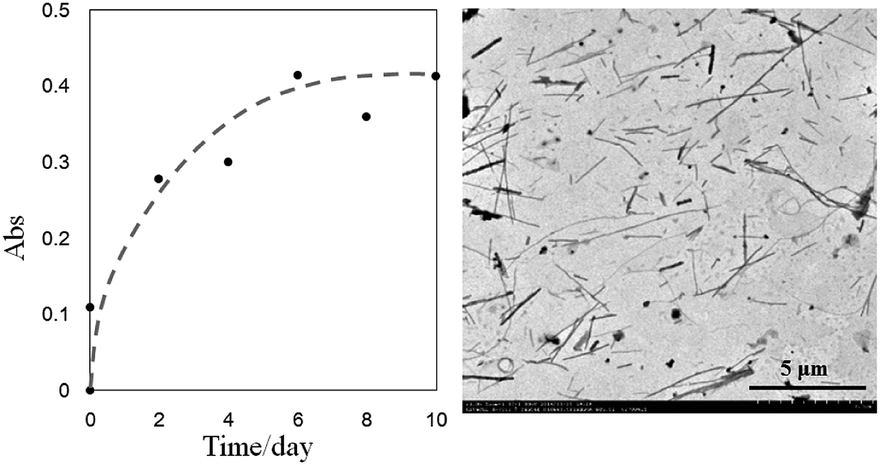 | ||
| Fig. 5 Changes of the UV-vis absorption (485 nm) of Dox moieties in the supernatant solution with time (Left). The collected precipitates were re-suspended in pH 7 water. The concentration of Dox group in the solution reached about 0.6 mg ml−1 after 10 days, as calculated according to the standard curve. The corresponding TEM image of the solution after 7 days (right). The scale bar is 5 μm. | ||
It should be noticed that for the re-suspended sample, not much non-fibrillar aggregates were observed even after 7 days at pH 7 and room temperature. Since un-conjugated Dox has low solubility at pH 7 and would precipitate in its own form, this suggested that not much drug was detached from the polymer during this incubation process. This was also supported by the 1H NMR, DOSY and UV-vis spectra of the dissociated fibrils (Fig. S6–S8†). In contrast, after 40 days of incubation, particle precipitates appeared on the bottom of the solution and the 1H NMR results suggested that they were mostly consisted of the detached Dox molecules (Fig. S9†).
Finally, we would like to study the possible structure of the fibril aggregates. The secondary structure of the aggregates were characterized with CD (Fig. 6). In the pH 7.6 aqueous solution, the PGA–Dox chains were negatively charged and adopted random coil structure. As pH decreased, the negative charges were gradually neutralized, and the chains took typical α-helix conformations at pH below 5.7 (Fig. 6A). Similar phenomena were observed with un-conjugated PGA.22,23 When heated to 60 °C, higher content of random coil structure were observed compared with that at the room temperature. Therefore, at the aggregate-forming condition in our study, the polymer–drug conjugates chain initially should take a mixed conformation with both α-helix and random coil components. As for the precipitates, the CD result also suggested that the PGA backbone took similar secondary structure of mixed random coil and α-helix components (Fig. 6B). It should be noted that many poly(amino acid)s tend to form β-sheet fibrillar structure under appropriate conditions,24–26 including the un-conjugated PGA, which would form amyloid-like fibrillar aggregates in acidic conditions (Fig. S10†).24,27 We initially thought that the fibril aggregates in our current system might possess amyloid-like structures, as many peptide–drug conjugates were proved to self-assembly into amyloid structure.28 However, the CD spectra of our samples showed no significant β-sheet content and dye binding experiments with thioflavin T and Congo red also showed negative results of the existence of amyloid structures (data not shown). Alternatively, there are evidences that Dox itself and other hydrophobic drugs would self-assemble into fibril-shaped aggregates under physiological condition.29–31 Therefore, it is likely that in our system, Dox moieties aggregate and form the hydrophobic core, while the PGA chains take mixed α-helix and random coil secondary structure and form the hydrophilic sheath of the fibrils.
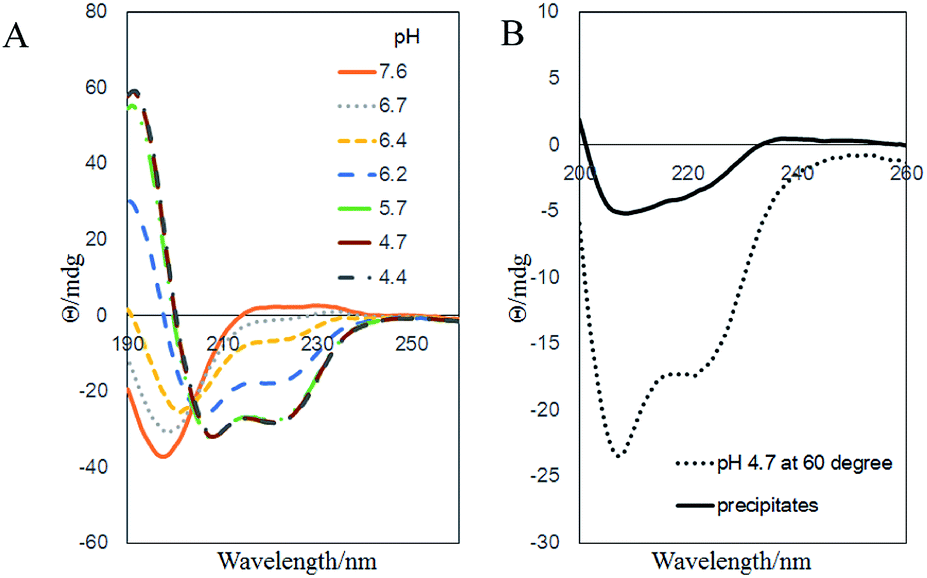 | ||
| Fig. 6 Far-UV CD spectra of (A) 0.5 mg ml−1 PGA–Dox solution at different pHs and room temperature, (B) the same sample at pH 4.7, 60 °C and the collected precipitates. | ||
Conclusions
In summary, we proved that the polymer–drug conjugate PGA–Dox can form fibrillar aggregates with diameter of about 40 nm and length up to tens of microns. Under the physiological condition (pH 7, room temperature), these aggregates dissolve slowly in days, i.e., seemingly useful as a sustained-releasing nano-fibril depot of polymer–drug conjugates. Through this model, we demonstrate a new strategy to decouple the particle size and the molecular weight by controlling the aggregation state of polymer–drug conjugates.It should be noted that the current study used an amide-linked drug–polymer conjugate, which is a stable linker and not much drugs should be detached from the polymer chain. Therefore, the current study only served to show the fibril aggregation can be formed in the drug–polyglutamate system. In our next step, a more relevant linker, such as the enzyme-cleavable oligopeptide linker,32 will be used to synthesize the conjugate, and then more studies on the drug release, serum stability, and the biological activity will be conducted.
Acknowledgements
The study is supported by Natural Science Foundation of China (NSFC Project no. 21434008 and 21074064).Notes and references
- H. Ringsdorf, J. Polym. Sci., Polym. Symp., 1975, 51, 135 CrossRef CAS PubMed.
- R. Duncan, Nat. Rev. Cancer, 2006, 6, 688 CrossRef CAS PubMed.
- J. Kopeček, Adv. Drug Delivery Rev., 2013, 65, 49 CrossRef PubMed.
- C. Li and S. Wallace, Adv. Drug Delivery Rev., 2008, 60, 886 CrossRef CAS PubMed.
- A. Bernareggi, C. Allievi, G. Pezzoni, A. Danese, and J. W. Singer, poster at The 46th AFI Symposium, Rimini, Italy, 2006 Search PubMed.
- D. G. Rudmann, J. T. Alston, J. C. Hanson and S. Heidel, Toxicol. Pathol., 2013, 41, 970 CrossRef CAS PubMed.
- K. Ulbrich, Č. Koňák, Z. Tuzar and J. Kopeček, Makromol. Chem., 1987, 188, 1261 CrossRef CAS PubMed.
- P. Deo, N. Deo, P. Somasundaran, S. Jockusch and N. J. Turro, J. Phys. Chem. B, 2005, 109, 20714 CrossRef CAS PubMed.
- J. G. Shiah, Č. Koňák, J. D. Spikes and J. Kopeček, J. Phys. Chem., 1997, 101, 6803 CrossRef CAS.
- A. Paul, M. J. Vicent and R. Duncan, Biomacromolecules, 2007, 8, 1573 CrossRef CAS PubMed.
- S. K. Filippov, J. M. Franklin, P. V. Konarev, P. Chytil, T. Etrych, A. Bogomolova, M. Dyakonova, C. M. Papadakis, A. Radulescu, K. Ulbrich, P. Stepanek and D. I. Svergun, Biomacromolecules, 2013, 14, 4061 CrossRef CAS PubMed.
- S. K. Filippov, P. Chytil, P. V. Konarev, M. Dyakonova, C. Papadakis, A. Zhigunov, J. Plestil, P. Stepanek, T. Etrych, K. Ulbrich and D. I. Svergun, Biomacromolecules, 2012, 13, 2594 CrossRef CAS PubMed.
- Y. J. Jun, J. H. Min, J. H. Yoo, J. H. Kim, H. J. Lee, B. Jeong and Y. S. Sohn, Bioorg. Med. Chem. Lett., 2008, 18, 6410 CrossRef CAS PubMed.
- S. Van, S. K. Das, X. Wang, Z. Feng, Y. Jin, Z. Hou, F. Chen, A. Pham, N. Jiang, S. B. Howell and L. Yu, Int. J. Nanomed., 2010, 5, 825 CrossRef CAS PubMed.
- H. Maeda, J. Wu, T. Sawa, Y. Matsumura and K. Hori, J. Controlled Release, 2000, 65, 271 CrossRef CAS.
- S. Li and L. Huang, Mol. Pharm., 2008, 5, 496 CrossRef CAS PubMed.
- L. Tang, X. Yang, Q. Yin, K. Cai, H. Wang, I. Chaudhury, C. Yao, Q. Zhou, M. Kwon, J. A. Hartman, I. T. Dobrucki, L. W. Dobrucki, L. B. Borst, S. Lezmi, W. G. Helferich, A. L. Ferguson, T. M. Fan and J. Cheng, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 15344 CrossRef CAS PubMed.
- Z. Luo and G. Zhang, J. Phys. Chem. B, 2009, 113, 12462 CrossRef CAS PubMed.
- W. A. R. van Heeswijk, C. J. T. Hoes, T. Stoffer, M. J. D. Eenink, W. Potman and J. Feijen, J. Controlled Release, 1985, 1, 301 CrossRef CAS.
- P. Changenet-Barret, T. Gustavsson, D. Markovitsi, I. Manet and S. Monti, Phys. Chem. Chem. Phys., 2013, 15, 2937 RSC.
- P. Agrawal, S. K. Barthwal and R. Barthwal, Eur. J. Med. Chem., 2009, 44, 1437 CrossRef CAS PubMed.
- E. J. Spek, Y. Gong and N. R. Kallenbach, J. Am. Chem. Soc., 1995, 117, 10773 CrossRef CAS.
- A. Holtzer and R. B. Hawkins, J. Am. Chem. Soc., 1996, 118, 4220 CrossRef CAS.
- M. Fändrich and C. M. Dobson, EMBO J., 2002, 21, 5682 CrossRef PubMed.
- J. Lai, C. Zheng, D. Liang and Y. Huang, Biomacromolecules, 2013, 14, 4515 CrossRef CAS PubMed.
- J. Lai, W. Fu, L. Zhu, R. Guo, D. Liang, Z. Li and Y. Huang, Langmuir, 2014, 30, 7221 CrossRef CAS PubMed.
- M. Colaco, J. Park and H. Blanch, Biophys. Chem., 2008, 136, 74 CrossRef CAS PubMed.
- R. Lin, A. G. Cheetham, P. Zhang, Y. A. Lin and H. Cui, Chem. Commun., 2013, 49, 4968 RSC.
- L. Zhu, S. Yang, X. Qu, F. Zhu, Y. Liang, F. Liang, Q. Wang, J. Li, Z. Li and Z. Yang, Polym. Chem., 2014, 5, 5700 RSC.
- L. Mao, H. Wang, M. Tan, L. Ou, D. Kong and Z. Yang, Chem. Commun., 2012, 48, 395 RSC.
- C. Yang, D. Li, Q. Zhao, L. Wang, L. Wang and Z. Yang, Org. Biomol. Chem., 2013, 11, 6946 CAS.
- R. Duncan, H. C. Cable, J. B. Lloyd, P. Rejmanová and J. Kopeček, Makromol. Chem., Rapid Commun., 1983, 184, 1997 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: GPC results, 1H NMR, DOSY, UV-vis, fluorescence spectra and TEM images. See DOI: 10.1039/c5ra06755c |
| This journal is © The Royal Society of Chemistry 2015 |