DOI:
10.1039/C5RA08733C
(Paper)
RSC Adv., 2015,
5, 48861-48867
Preparation of 2-phenyl-3-hydroxyquinoline-4(1H)-one-5-carboxamides as potential anticancer and fluorescence agents†
Received
11th May 2015
, Accepted 28th May 2015
First published on 28th May 2015
Abstract
The synthesis of 3-hydroxyquinoline-4(1H)-one derivatives bearing substituted phenyl in position 2 and variously substituted carboxamide group in position 5 is described, with use of 3-nitrophthalic anhydride, α-haloketones and primary amines as the starting materials. The synthetic approach was inspired by the preparation of analogous derivatives reported previously. However, a different strategy had to be developed with the corresponding bis(phenacyl)-3-aminophthalates as the key intermediates. Synthesized hydroxyquinolinones, as well as their intermediates, were tested for their cytotoxic activity towards various cancer and non-malignant cell lines. The fluorescent properties of these compounds have also been evaluated. In both fields, interesting data were obtained and compared to isomeric compounds that have been studied in the past.
Introduction
In the last decade, compounds bearing 3-hydroxyquinolin-4(1H)-one scaffold (3HQs) have been studied intensively due to their outstanding biological and spectral properties. In the field of medicinal chemistry, 3HQs exhibited significant cytotoxicity against selected cancer cell lines, indicating their possible application as novel anticancer agents.1–5 Consequently, the most promising compounds have been submitted for liposomal solubilization6 and micellar dispersion7 studies to increase their bioavailability and stability. Aside from cytotoxicity, hydroxyquinolinones also exhibited strong fluorescence with a dual emission spectra which pointed to their potential application in the area of molecular probes and fluorescence labeling.8–11 With use of the highly efficient method of preparation, discovered in the end of nineties,12–16 a number of variously substituted 3HQs have been synthesized and studied. Recently, a deeper attention has been paid to compounds bearing the carboxamide group located on a benzene ring of the quinolinone scaffold. For such compounds, a high-throughput solid-phase synthesis concept has been developed17–19 and targeted chemical libraries of 3HQs with the carboxamide group located in position 6, 7 and 8 were prepared. Both structure–cytotoxicity20,21 and structure–fluorescence22 relationships have been evaluated and derivatives with the most promising biological and fluorescence properties were identified (Fig. 1).
 |
| | Fig. 1 Solid-phase strategy for the preparation of 6-8 carboxamides described previously17,20 and properties of selected compounds. | |
Despite detailed research into this field, a aforementioned studies were incomplete due to the impossibility to apply the original solid-phase approach to the preparation of isomers with the carboxamide group located in position 5. To reach this goal, we switched to traditional solution-phase chemistry to find a synthetic strategy for the desired compounds. Herein we describe the method development, its application for the preparation of a set of target derivatives and comparisons of their cytotoxic/fluorescence properties with the corresponding 6-8-carboxamides.
Results and discussion
Synthesis
The initial strategy (Scheme 1) was based on the method of preparation of the corresponding 6-8 carboxamides reported previously.17 In such cases the key building block was 3-amino-2-(methoxycarbonyl)benzoic acid 3. We managed to synthesize the compound from 3-nitrophtalic anhydride 1 via intermediate 2 according to described procedures.23,24 However, the crude purity of compound 3 after the catalytic reduction step was low. Conversion of carboxylic acid 3 to the corresponding amide was tested with propylamine. Unfortunately, the attempt to synthesize compound 4 with use of the DIC/HOBt method afforded the product, only as a part of a complex mixture. We subsequently reversed the reaction sequence and propylamine was acylated directly with compound 2 via the corresponding acylchloride. In this case, the expected product was not obtained and the reaction afforded pure 4-nitro-2-propylisoindoline-1,3-dione 5. Although we managed to hydrolyze the intermediate 5 to yield 2-nitro-6-(propylcarbamoyl)benzoic acid 6, its catalytic hydrogenation led only to a mixture of compounds and the desired product 7 was not isolated. Additionally, alkylation of acid 6 with bromoacetophenone did not lead to the corresponding phenacylester (which could be also applied for the preparation of target 3HQs) but intermediate 5 was detected instead.
 |
| | Scheme 1 Attempt to synthesize methyl 2-amino-6-(propylcarbamoyl)benzoate 7 according to previously reported procedure. | |
The alternative strategy was based on the preparation of bis(phenacyl)-3-aminophthalates 13 as the precursors for the formation of the 3HQ scaffold. Due to limited purity after catalytic reduction of compound 2, dimethylester 8 was synthesized first, via the formation of the corresponding acylchloride. Subsequent reduction afforded compound 9 in an excellent purity which was smoothly hydrolyzed to obtain pure 3-aminophthalic acid 10. Compound 10 was alkylated with bromoacetophenone to give bis(phenacyl)-3-aminophtalate 13a. It is worth mentioning that an alternative strategy based on the hydrolysis of compound 8 followed by alkylation of intermediate 11 failed in the stage of catalytic hydrogenation of compound 12, which afforded only a complex mixture of compounds (Scheme 2). Cyclization of intermediate 13a to 3HQ derivative 14a was performed with use of TFA as described earlier for similar derivatives.12 The final step of the sequence was aminolysis of compound 14a with propylamine to obtain final 3HQ 15a. Two methods have been developed: reflux with propylamine in chloroform (10% solution) was successfully used on a small scale (milligram quantities). On a larger scale (quantities above 100 mg), the method was not successful due to an incomplete conversion and chloroform had to be replaced by DMF.
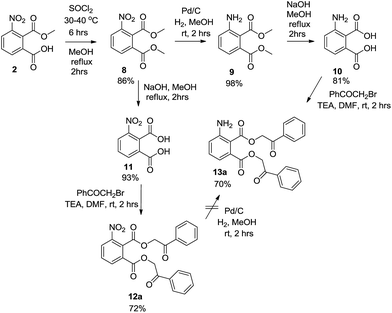 |
| | Scheme 2 Strategy for the preparation of bis(phenacyl)-3-aminophthalates 13a. | |
For fluorescence and biological assay purposes we also managed to develop re-esterification of compound 14a to prepare methyl derivative 16a (Scheme 3).
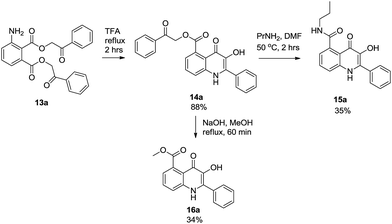 |
| | Scheme 3 Conversion of intermediate 13a to hydroxyquinolinone derivative 15a | |
With use of developed procedures, a set of model compounds (Table 1) were prepared from various bromoacetophenones and amines (Fig. 2). To allow for the comparative study of the structure–cytotoxicity and structure–fluorescence relationship, we selected the same building blocks and their combinations as used previously for the preparation of 6-8 carboxamides.20,21 The developed procedure was generally applicable, only the cyclization of intermediate 13h had to be performed in anhydrous phosphoric acid instead of TFA which did not work.
Table 1 List of synthesized 3HQs and bis(phenacyl)-3-aminophthalates
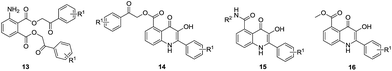
|
| Entry |
Cmpd |
R1 |
R2 |
Yielda (%) |
| Calculated after the cyclization step. |
| 1 |
13a |
H |
— |
70 |
| 2 |
13b |
3,5-DiCl-4-NH2 |
— |
68 |
| 3 |
13c |
4-CH3 |
— |
84 |
| 4 |
13d |
4-OCH3 |
— |
65 |
| 5 |
13e |
4-F |
— |
82 |
| 6 |
13f |
3-Br |
— |
77 |
| 7 |
13g |
3-NO2-4-Cl |
— |
68 |
| 8 |
13h |
3-NO2-4-N-PIP |
— |
87 |
| 9 |
14a |
H |
— |
88 |
| 10 |
14b |
3,5-DiCl-4-NH2 |
— |
87 |
| 11 |
14c |
4-CH3 |
— |
93 |
| 12 |
14d |
4-OCH3 |
— |
84 |
| 13 |
14e |
4-F |
— |
90 |
| 14 |
14f |
3-Br |
— |
95 |
| 15 |
14g |
3-NO2-4-Cl |
— |
81 |
| 16 |
14h |
3-NO2-4-N-PIP |
— |
79 |
| 17 |
15a |
H |
Propyl |
35 |
| 18 |
15b |
3,5-DiCl-4-NH2 |
Propyl |
45 |
| 19 |
15c |
4-CH3 |
Propyl |
64 |
| 20 |
15d |
4-OCH3 |
Propyl |
40 |
| 21 |
15e |
4-F |
Propyl |
31 |
| 22 |
15f |
3-Br |
Propyl |
81 |
| 23 |
15g |
3,5-DiCl-4-NH2 |
Hydroxyethyl |
35 |
| 24 |
15h |
3,5-DiCl-4-NH2 |
 |
24 |
| 25 |
15i |
3,5-DiCl-4-NH2 |
Benzyl |
34 |
| 26 |
15j |
3,5-DiCl-4-NH2 |
H |
30 |
| 27 |
16a |
H |
— |
34 |
| 28 |
16b |
3,5-DiCl-4-NH2 |
— |
32 |
 |
| | Fig. 2 List of used haloketones and amines. | |
Fluorescence properties
2-Aryl-3-hydroxyquinolin-4(1H)-ones typically exhibit dual fluorescence spectrum with two sufficiently separated emission bands, which allows for their possible application as fluorescent labels.10,11 Type/location of different substituents on the 3HQ scaffold can significantly affect the emission spectra. In the case of different isomeric 3HQ-carboxamides, we have already reported that positioning of the carboxamide group strongly influences the resulting fluorescence properties: 6-carboxamides exhibited two well separated maxima whereas 8-carboxamides provided only one emission maximum at lower wavelength and extremely low quantum yields.22
The difference could be explained by the strong intramolecular hydrogen bonding in the structure of 8-carboxamides which does not exist in the case of the 6- and 7-carboxamide analogues (Fig. 3).
 |
| | Fig. 3 Different sterical features of isomeric 3HQ-carboxamides. | |
A different situation was expected in the structure of 5-carboxamides; the steric repulsion between two carbonyl groups enforces the conformation in which the carboxamide is not in the same plain with the aromatic moiety and conjugation is lost. This fact has been proved by X-ray analysis of compound 15a (Chart 1). However, the consequence of this feature to the resulting fluorescence and biological properties was unclear.
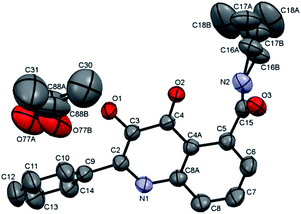 |
| | Chart 1 X-ray analysis of compound 15a. Hydrogen atoms have been omitted for the sake of clarity. Displacement ellipsoids are drawn at the 50% probability level. | |
From the initial results of the fluorescence study in dimethyl sulfoxide (DMSO), it was evident that the existence of the dual fluorescence spectrum was not significantly affected by the location of the carboxamide group at position 5. Although the intensity of the lower wavelength maximum was reduced (Fig. 4, solid line), it was still noticeable and resembled the emission spectra of 7-carboxamides.22 On the other hand, the presence of the methyl ester instead of carboxamide (3HQs 16a, 16b) led to complete loss of the emission maximum at lower wavelength and the dual fluorescence spectrum was not detected. 3HQs 14 with the phenacyl ester moiety did not give consistent results: for the majority of them the dual fluorescence spectrum was lost, only two 3HQs 14a and 14f provided the second (higher wavelength) emission maximum. In contrast, 3HQ 14g afforded only the lower wavelength emission maximum (Table 2) and compound 14h did not exhibit any fluorescence at all. To highlight the key effect of the 3HQ scaffold, we have also studied bis(phenacyl)esters 13. Although their fluorescence has been observed, the dual fluorescence spectra were not obtained and their emission maximum was significantly lower compared to 3HQs (520–560 nm for 3HQs 14–16, 454–480 for compounds 13).
 |
| | Fig. 4 Excitation and emission spectra for 13c (dash line), 15d (solid line) and 16a (dotted line). | |
Table 2 Fluorescence properties of synthesized compounds (measured in dimethyl sulfoxide, conc. 0.1 mg mL−1)
| Entry |
Cmpd |
λexca (nm) |
λem,1b (nm) |
λem,2c (nm) |
I1/I2d |
φe (%) |
| λex, excitation wavelength. λem,1, the fluorescence emission maximum at lower wavelengths. λem,2, the fluorescence emission maximum at higher wavelengths. I1/I2, the ratio of fluorescence maxima intensities. φ, fluorescence quantum yield (determined with quinine sulphate in 0.5 M sulphuric acid (φ = 0.577 (ref. 25)), taken as a reference fluorescence standard). |
| 1 |
13a |
394 |
474 |
— |
— |
43.43 |
| 2 |
13b |
373 |
467 |
— |
— |
16.65 |
| 3 |
13c |
390 |
464 |
— |
— |
48.67 |
| 4 |
13d |
371 |
454 |
— |
— |
20.15 |
| 5 |
13e |
373 |
462 |
— |
— |
3.66 |
| 6 |
13f |
394 |
474 |
— |
— |
19.87 |
| 7 |
13g |
394 |
472 |
— |
— |
5.43 |
| 8 |
13h |
395 |
480 |
— |
— |
0.17 |
| 9 |
14a |
402 |
474 |
555 |
0.1158 |
11.20 |
| 10 |
14b |
413 |
— |
560 |
— |
19.57 |
| 11 |
14c |
405 |
— |
555 |
— |
10.44 |
| 12 |
14d |
403 |
— |
560 |
— |
28.91 |
| 13 |
14e |
404 |
— |
556 |
— |
10.84 |
| 14 |
14f |
407 |
478 |
555 |
0.0990 |
7.26 |
| 15 |
14g |
419 |
481 |
— |
— |
0.72 |
| 16 |
14h |
— |
— |
— |
— |
— |
| 17 |
15a |
403 |
449 |
527 |
0.1024 |
36.14 |
| 18 |
15b |
409 |
467 |
531 |
0.0627 |
37.63 |
| 19 |
15c |
404 |
464 |
526 |
0.1025 |
14.76 |
| 20 |
15d |
400 |
454 |
525 |
0.1146 |
38.61 |
| 21 |
15e |
404 |
455 |
528 |
0.1123 |
42.54 |
| 22 |
15f |
403 |
457 |
520 |
0.0698 |
30.80 |
| 23 |
15g |
415 |
456 |
532 |
0.0602 |
42.74 |
| 24 |
15h |
411 |
468 |
532 |
0.0893 |
24.11 |
| 25 |
15i |
410 |
461 |
531 |
0.0683 |
37.10 |
| 26 |
15j |
412 |
471 |
530 |
0.0855 |
25.51 |
| 27 |
16a |
402 |
— |
547 |
— |
39.78 |
| 28 |
16b |
416 |
— |
554 |
— |
29.96 |
Quantum yields within the group of 5-carboxamides 15 were mostly similar, although slightly lower compared to analogical 6/7-carboxamides.22 Nonetheless, there was no clear relationship between their values and the structural features of these molecules. A quite different situation was observed for phenacyl-3HQs 14 which provided a wide range of values from 0.72% to 28.9%. It could be concluded that the presence of 2-phenyl with strongly electron withdrawing groups (such as nitro, fluoro) significantly diminished the quantum yields (compounds 14g and 14h), whereas compounds with electron-donating groups (such as methoxy, amino, methyl) provided higher values. Similar dependence have been observed for bis(phenacyl)esters 13.
The study was further expanded to evaluate the relationship between fluorescence properties and pH. For this purpose, representative compounds 13c, 14a, 15d and 16a with the highest quantum yields and/or dual-band spectra were selected. The pH measurements were performed in a solution consisting of 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v 0.1 M phosphate buffer–dimethyl sulfoxide with the concentration of investigated compound being 10 μg mL−1. Surprisingly, in aqueous solution the emission spectra totally lost their dual character. The loss of the dual shape of emission spectra did not enable us to apply the ratio of the maximum intensities as a signal. The intensity ratio does not usually depend on the label concentration which is an important advantage in complex biological systems, such as cells or tissues, where the local concentration of the dye cannot be easily controlled and generally the label is not distributed homogenously. In each studied case, the emission spectra retained the same shape at different pHs, whereas the fluorescence intensity changed (Fig. 5). The maximum fluorescence intensity was reached at pH 3.60, 6.41, 5.23 and 5.75 for 13c, 14a, 15d and 16a, respectively. The pH dependences of fluorescence intensity had a similar shape for all investigated compounds: at acidic pH values the fluorescence intensity increased with increasing pH, reached the maximum and then gradually decreased (Fig. 5, below). In spite of the fact that the ratiometric measurement was not possible, the pH dependence of fluorescence intensity for 13c was linear (y = −8.7349x + 132.15, R2 = 0.9936) in the pH range 4.62–9.22 which could be beneficial, especially for biological measurement.
:1 v/v 0.1 M phosphate buffer–dimethyl sulfoxide with the concentration of investigated compound being 10 μg mL−1. Surprisingly, in aqueous solution the emission spectra totally lost their dual character. The loss of the dual shape of emission spectra did not enable us to apply the ratio of the maximum intensities as a signal. The intensity ratio does not usually depend on the label concentration which is an important advantage in complex biological systems, such as cells or tissues, where the local concentration of the dye cannot be easily controlled and generally the label is not distributed homogenously. In each studied case, the emission spectra retained the same shape at different pHs, whereas the fluorescence intensity changed (Fig. 5). The maximum fluorescence intensity was reached at pH 3.60, 6.41, 5.23 and 5.75 for 13c, 14a, 15d and 16a, respectively. The pH dependences of fluorescence intensity had a similar shape for all investigated compounds: at acidic pH values the fluorescence intensity increased with increasing pH, reached the maximum and then gradually decreased (Fig. 5, below). In spite of the fact that the ratiometric measurement was not possible, the pH dependence of fluorescence intensity for 13c was linear (y = −8.7349x + 132.15, R2 = 0.9936) in the pH range 4.62–9.22 which could be beneficial, especially for biological measurement.
 |
| | Fig. 5 Illustrative emission spectra of 13c (red, solid line pH 5.75, dashed line pH 7.69 and dotted line pH 12.49), 14a (green, solid line pH 6.43, dashed line pH 2.48 and dotted line pH 11.53), 15d (black, solid line pH 3.60, dashed line pH 6.49 and dotted line pH 12.49) and 16a (blue, solid line pH 5.75, dashed line pH 8.58 and dotted line pH 11.63) and fluorescence intensity at different pH. | |
Cytotoxic activity
To allow for the structure–activity relationship studies, the synthesized compounds were screened against the same cancer cell lines as their analogues, that have previously been studied: CCRF-CEM (T-lymphoblastic leukaemia), CEM-DNR (T-lymphoblastic leukaemia, daunorubicin resistant), K562 (acute myeloid leukaemia) and K562-TAX (acute myeloid leukaemia, paclitaxel resistant), A549 (human lung adenocarcinoma), HCT116 (human colorectal cancer), HCT116p53−/− (human colorectal cancer, p53 deficient), U2OS (human osteosarcoma). The therapeutic index (TI) was evaluated using one representative of non-malignant cell lines – BJ (human fibroblasts). The obtained results are summarized as the IC50 values in Table 3.
Table 3 Results of cytotoxicity for all synthesized compounds (relative IC50, μM). Average values from 3–4 independent experiments with SD ranging from 10–25% of the average values
| Cmpd |
CCRF-CEM |
CEM-DNR |
K562 |
K562-TAX |
A549 |
HCT116 |
HCT116 p53−/− |
U2OS |
BJ |
| 13a |
>50 |
>50 |
>50 |
>50 |
>50 |
43 |
>50 |
>50 |
>50 |
| 13b |
>50 |
48 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
| 13c |
>50 |
46 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
| 13d |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
| 13e |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
| 13g |
12 |
37 |
17 |
8.3 |
>50 |
30 |
19 |
12 |
>50 |
| 13h |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
>50 |
| 14a |
2.7 |
8.1 |
8.6 |
6.0 |
9.5 |
12 |
4.0 |
10 |
10 |
| 14b |
0.8 |
4.5 |
1.5 |
0.8 |
2.3 |
1.6 |
1.4 |
2.4 |
25 |
| 14c |
40 |
>50 |
>50 |
42 |
>50 |
>50 |
>50 |
>50 |
>50 |
| 14d |
2.7 |
8.8 |
7.6 |
4.2 |
13 |
6.5 |
4.0 |
10 |
>50 |
| 14e |
2.2 |
6.7 |
13 |
4.0 |
23 |
3.5 |
3.3 |
5.4 |
36 |
| 14f |
2.3 |
2.3 |
1.3 |
4.6 |
6.4 |
3.0 |
2.7 |
10 |
34 |
| 14g |
20 |
24 |
38 |
6.2 |
12 |
8.7 |
11 |
7.7 |
26 |
| 14h |
32 |
>50 |
>50 |
13 |
>50 |
12 |
32 |
>50 |
>50 |
| 15a |
10 |
26 |
>50 |
31 |
>50 |
20 |
19 |
23 |
>50 |
| 15b |
7.7 |
23 |
15 |
8.8 |
34 |
7.5 |
7.2 |
12 |
34 |
| 15c |
22 |
39 |
32 |
26 |
>50 |
27 |
20 |
>50 |
>50 |
| 15d |
28 |
44 |
48 |
35 |
>50 |
30 |
27 |
>50 |
>50 |
| 15e |
16 |
36 |
>50 |
32 |
>50 |
21 |
17 |
46 |
45 |
| 15f |
4.3 |
13 |
25 |
10 |
24 |
7.9 |
8.0 |
14 |
>50 |
| 15g |
31 |
>50 |
49 |
48 |
>50 |
42 |
39 |
47 |
>50 |
| 15h |
34 |
>50 |
36 |
>50 |
48 |
25 |
27 |
46 |
40 |
| 15i |
3.3 |
10 |
2.5 |
4.6 |
6.9 |
24 |
4.0 |
10 |
8.6 |
| 15j |
34 |
48 |
45 |
30 |
49 |
20 |
18 |
27 |
45 |
| 16a |
15 |
49 |
>50 |
>50 |
>50 |
>50 |
46 |
17 |
>50 |
| 16b |
4.7 |
6.7 |
5.8 |
4.6 |
27 |
5.5 |
4.6 |
9.3 |
9.8 |
As in the case of bisphenacyl-2-aminoterephthalates,2 the corresponding bisphenacyl-3-aminophthalates 13 did not exhibit significant cytotoxic activity. However, an exception has been observed for compound 13g which showed a medium cytotoxicity against K562-TAX, U2OS and CEM cells. Within the group of HQ-5-carboxamides 15, the similar SAR pattern as for analogical 6-8 carboxamides was observed: the highest activity was detected for 2-(3,5-dichloro-4-aminophenyl)-3HQs with unpolar N-alkyl substituents (15b:N-propyl and 15i:N-benzyl), whereas the unsubstituted carboxamide 15j and compounds with polar (15g:N-hydroxyethyl) or basic (15h:N-piperidinyl-ethyl) ligands were not cytotoxic. Structural change of propylamides (15a,b) to the corresponding methylesters (16a,b) provided approximately the same results. On the other hand, cytotoxicity of 3HQs-5-phenacylesters 14 was in general significantly higher, with compound 14b being the most active derivative from the whole set. IC50 of aminophthalate 14b for A-549, K56A, CEM, K562-TAX and CEM-DNR was comparable to isomeric aminoterephthalate.2 In contrast, 4-methylphenyl phthalate derivative 14c was inactive compared to isomeric aminoterephthalate that exhibited micromolar IC50 for all tested lines.2 The reversed dependence was observed for the unsubstituted phenacylester 14a (Fig. 6). The majority of active compounds were less active against the CEM-DNR multidrug resistant cell line overexpressing the multidrug resistance protein 1 (MRP-1), than against highly chemosensitive parental CCRF-CEM cell line. However, the opposite pattern was identified in the case of P glycoprotein (Pgp-1) overexpressing multidrug resistant K562-TAX cell line which was more sensitive than parental K562 cell line. Interestingly, CEM-DNR cells also lack topoisomerase IIα gene, which has been previously reported as a molecular target for quinolone derivatives. It is suggesting either the topoisomerase IIα as a molecular target for 3HQs-5-phenacylesters 14 or involvement of MPR-1 but not Pgp-1 in drug efflux and resistance mechanisms.26 The growth inhibitory activity of compounds against human colorectal cancer cell line HCT116 and its p53 deficient counterpart (HCT116p53−/−) were similar, thus indicating independence of cell death mechanism on the p53 gene. The therapeutic index of the most active compound 14b ranged from 15–30 for majority of cell lines, suggesting preferential activity against malignant cells.
 |
| | Fig. 6 Example of 3HQs-phenacylesters cytotoxicity dependence on the location of the ester group (for K562 cell line). | |
Finally, two representative compounds were subjected to the fluorescent microscopy assay. We selected the most cytotoxic compound 14b along with the most active carboxamide derivative 15i. The results are depicted in Chart 2. The microscopy imaging is showing that the fluorescent compounds are penetrating cellular membranes of living cells. The highest intensity of the fluorescence and accumulation of the compound was observed in the cytoplasm, with discrete nuclear spots. Maximum cytoplasmic positivity was seen in perinuclear region, the staining pattern was overlapping in both emission wavelengths. Detailed subcellular localization is to be determined in future studies.
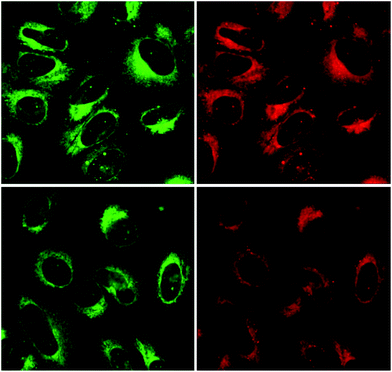 |
| | Chart 2 U2OS osteosarcoma cancer cells treated with 14b (first row) and 15i (second row). Full size images are available in ESI.† | |
Conclusions
We have developed a synthetic procedure for 3HQs-5-carboxamides 15 to enable the study of their properties compared to analogical 6-8 isomers. In the field of fluorescence properties, the synthesized 5-carboxamides exhibited similar results to their 7-isomers that have previously been studied. In this regard, some derivatives showed a potential to serve as pH indicators and/or molecular probes. Based on the fluorescence microscopy imaging of U2OS cells treated with two representative compounds 14b and 15i it was demonstrated, that compounds enter live cells and show preferential cytoplasmic staining. The cytotoxicity of 3HQs-5-carboxamides towards various cancer cell lines corresponded to 8-carboxamides. The overall results show that the location of the carboxamide group in position 5 rather decreases the cytotoxicity activity compared to 6,7-carboxamides, however, the evaluation of SAR data revealed the same pattern. Higher cytotoxicity was detected for 3HQs-5-phenacylesters 14. The most active compound 14b exhibited the similar activity as the corresponding 7-phenacylester described previously. On the other hand, the SAR pattern observed within the group of available 5- and 7-phenacylesters showed an interesting differences based on the location of the phenacylester moiety. To explore the field in further details, 3HQs-6-phenacylesters and 3HQs-8-phenacylesters will be synthesized and systematically studied for biological activities and fluorescence probing properties.
Acknowledgements
The authors are grateful to projects CZ.1.07/2.3.00/30.0060, CZ.1.07/2.3.00/30.0041 from the European Social Fund, from Palacky University (Internal Grant no. IGA_PrF_2015_007, IGA_LF_2015_031), National Sustainability Programme (LO1304) and Czech Technology Agency (TE02000058).
Notes and references
- P. Hradil, P. Krejčí, J. Hlaváč, I. Wiedermannová, A. Lyčka and V. Bertolasi, J. Heterocycl. Chem., 2004, 41, 375–379 CrossRef CAS PubMed.
- M. Soural, J. Hlaváč, P. Hradil, I. Fryšová, M. Hajdúch, V. Bertolasi and M. Maloň, Eur. J. Med. Chem., 2006, 41, 467–474 CrossRef CAS PubMed.
- P. Hradil, J. Hlaváč, M. Soural, M. Hajdúch, M. Kolář and R. Večeřová, Mini-Rev. Med. Chem., 2009, 9, 696–702 CrossRef CAS.
- P. Krejčí, P. Hradil, J. Hlaváč and M. Hajdúch, WO2008028427 A1, 2008.
- Z. Sui, V. N. Nguyen, J. Altom, J. Fernandez, J. J. Hilliard, J. I. Bernstein, J. F. Barrett and K. A. Ohemeng, Eur. J. Med. Chem., 1999, 34, 381–387 CrossRef CAS.
- M. Di Cagno, P. C. Stein, J. Stýskala, J. Hlaváč, N. Skalko-Basnet and A. Brauer-Brandl, Eur. J. Pharm. Biopharm., 2012, 80, 657–662 CrossRef CAS PubMed.
- M. Di Cagno, J. Stýskala, J. Hlaváč, M. Brandl, A. Brauer-Brandl and N. Skalko-Basnet, J. Liposome Res., 2011, 21, 272–278 CrossRef CAS PubMed.
- K. Motyka, B. Vankova, J. Hlaváč and M. Soural, J. Fluoresc., 2011, 21, 2207–2212 CrossRef CAS PubMed.
- K. Motyka, J. Hlaváč, M. Soural, P. Hradil, P. Krejčí, L. Kvapil and M. Weiss, Tetrahedron Lett., 2011, 52, 715–717 CrossRef CAS PubMed.
- D. A. Yushchenko, V. V. Shvadchak, A. S. Klymchenko, G. Duportail, Y. Mely and V. G. Pivovarenko, New J. Chem., 2006, 30, 774–781 RSC.
- D. A. Yushchenko, M. D. Bilokiń, O. V. Pyvovarenko, G. Duportail, Y. Mely and V. G. Pivovarenko, Tetrahedron Lett., 2006, 47, 905–908 CrossRef CAS PubMed.
- M. Soural, P. Hradil, S. Křupková and J. Hlaváč, Mini-Rev. Org. Chem., 2012, 9, 426–432 CrossRef CAS.
- P. Hradil and J. Jirman, Collect. Czech. Chem. Commun., 1995, 60, 1357–1366 CrossRef CAS.
- P. Hradil, J. Vanecek, J. Hlavac and J. Sevcik, Collect. Czech. Chem. Commun., 1999, 64, 257–264 CrossRef CAS.
- P. Hradil, J. Hlaváč, P. Krejčí and K. Lemr, Acta Univ. Palacki. Olomuc., 1999, 38, 17–23 CAS.
- P. Hradil, J. Hlaváč and K. Lemr, J. Heterocycl. Chem., 1999, 36, 141–144 CrossRef CAS PubMed.
- M. Soural and V. Krchnak, J. Comb. Chem., 2007, 9, 793–796 CrossRef CAS PubMed.
- B. Vankova, J. Hlavac and M. Soural, J. Comb. Chem., 2010, 12, 890–894 CrossRef CAS PubMed.
- S. Krupkova, M. Soural, J. Hlavac and P. Hradil, J. Comb. Chem., 2009, 11, 951–955 CrossRef CAS PubMed.
- M. Soural, J. Hlavac, P. Funk, P. Dzubak and M. Hajduch, ACS Comb. Sci., 2011, 13, 39–44 CrossRef CAS PubMed.
- J. Kadric, K. Motyka, P. Džubák, M. Hajdúch and M. Soural, Tetrahedron Lett., 2014, 55, 3592–3595 CrossRef CAS PubMed.
- K. Motyka, J. Hlaváč, M. Soural and P. Funk, Tetrahedron Lett., 2010, 51, 5060–5063 CrossRef CAS PubMed.
- L. Marinus, Eur. Pat. Appl., 133310, 20 Feb, 1985.
- C. Banzatti, N. Carfagna, R. Commisso, F. Heidempergher, L. Pegrassi and P. Melloni, J. Med. Chem., 1988, 31, 1466–1471 CrossRef CAS.
- W. H. Melhuish, J. Phys. Chem., 1961, 65, 229–235 CrossRef CAS.
- V. Noskova, P. Dzubak, G. Kuzmina, A. Ludkova, D. Stehlik, R. Trojanec, A. Janostakova, G. Korinkova, V. Mihal and M. Hajduch, Neoplasma, 2002, 49, 418–425 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1057862. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c5ra08733c |
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 












