DOI:
10.1039/C5RA06728F
(Paper)
RSC Adv., 2015,
5, 64682-64688
A nano Ag5 cluster tip probing the vertical transfer of CO(ads) adsorbed on Ag(110) with simulated inelastic electron tunneling spectroscopy
Received
14th April 2015
, Accepted 21st July 2015
First published on 22nd July 2015
Abstract
A nano Ag5 cluster tip probing the vertical transfer of CO(ads) adsorbed on a Ag(110) surface has been investigated with simulated inelastic electron tunneling spectroscopy (IETS) generated by combining DFT-based molecular dynamic simulations with a Fourier transform of auto-correlation function of the derivative of local density of states (FT-ACF-δLDOS). It is found that tunneling conductance generated based on the trajectories of LDOS is significantly increased as a nano Ag5 cluster tip is introduced and their vibrational amplitudes of low-frequency modes are enhanced in IETS. In addition, the IETS shows the doublet feature in the regions of low-frequency mode, i.e. frustrated-rotation, and high-frequency mode, i.e. C–O stretching, respectively, due to the change of geometry of CO(ads) leading to the transfer of CO(ads) adsorbed on the Ag(110) surface to a nano Ag5 cluster tip. Furthermore, an anharmonic coupling between frustrated-rotation and C–O stretching mode is investigated by using time-resolved IETS analysis. Finally, the key issue regarding the activation of low-frequency modes by a nano Ag5 cluster tip to cause the transfer of CO(ads) adsorbed on Ag(110) surface is addressed.
Introduction
For more than two decades, a scanning tunneling microscopy (STM) tip1,2 has been used to position molecules on metal surfaces and to probe their electronic properties on an atomic scale. In addition, the tunneling electrons from an STM tip can be used as a non-thermal energy source to induce electronic vibrational excitation of adsorbates that cause various controllable surface chemical reactions, such as vertical manipulation,3,4 rotation,5,6 dehydrogenation,7,8 or lateral hopping process.9,10 The corresponding inelastic electron tunneling spectroscopy (IETS)11 can measure the change in tunneling conductance as electrons excite vibrations and is a powerful technique for the identification of vibrational signatures of single chemisorbed species on metal surfaces to predict and understand both bond-weakening and bond-breaking processes. In 2001, Ho et al.12 successfully demonstrated a tip-induced vertical transfer of CO(ads) adsorbed on the Ag(110) surface by combining both STM topological image and IETS measurement, that is, the CO(ads) is vertically transferred from the Ag(110) surface to the tip by dosing the electrons into molecule. In 2002, Persson13 followed the same strategy to investigate the lateral hopping process for CO(ads) adsorbed on the Pd(110) surface and it was found that its hopping rate is strongly related to intramolecular high-frequency stretching mode in IETS. Additionally, they believed that both vertical and lateral translations for the CO(ads) adsorbed on surface are induced by an anharmonic coupling between low-frequency modes (frustrated-translation and frustrated-rotation) and high-frequency vibrational mode (approximately 240 meV, C–O stretching mode).
Over past few years, theoretical research works have been devoted to developing the new methodology to simulate STM images and vibrational inelastic electron tunneling for providing more insights into the nature of the electron-vibration coupling in tunneling-current-induced reaction process.14–18 Luo and co-workers19 successfully generated IETS for CO(ads) adsorbed on Cu(100) based on Tersoff–Hamann (TH) approximation,20,21 in which the tunneling conductance is only proportional to local density of states (LDOS) at near Fermi level. In addition, G. Kirzenow's research group have generated the simulated IETS for identifying vibrational signatures of the propanedithiolate (PDT) molecule in Gold–thiol molecular junction and provide a good comparison with the experiment IETS results.22–24 More recently, an approach beyond TH approximation, which includes electron–phonon coupling effect, has been successful to calculate the IETS for atomic-gold wires, molecular junction and single molecule adsorbed on metal surfaces by using nonequilibrium Green's functions (NEGF).25–28 In addition, Flipse and co-workers simulated CO(ads) on Cu(100) including tip effect to demonstrate how chemically functionalized STM tips modify the IETS intensity with respect to their vibrational modes.29,30 Although these simulated IETS successfully provide a better understanding of electron tunneling process for CO(ads) adsorbed on the Cu(100) surface, the detailed reaction mechanism of tunneling-current-induced vertical transfer of CO(ads) adsorbed on the Ag(110) surface is still lacking due to the fact that these calculated IETS are generated based on static approach with harmonic approximation. Consequently, the effects of both temperature and tip on adsorption dynamics should be taken into account in the calculation to realize an anharmonic coupling for the adsorbate–surface system. Therefore, in this theoretical study, we will present a novel computational scheme to simulate IETS of CO(ads) adsorbed on the Ag(110) surface at finite temperature by using DFT-based molecular dynamic simulations in combination with a Fourier transform of auto-correlation function of the derivative of local density of states (FT-ACF-δLDOS). Furthermore, a nano Ag5 cluster tip is introduced to accurately describe the interaction between a tip and the sample. In addition, the effect of a tip on adsorption dynamics of CO(ads) adsorbed on the Ag(110) surface is investigated to understand the correlation between the orientation of CO(ads) molecule axis and its tunneling conductance during dynamic simulation at finite temperature. Finally, we will explore the corresponding anharmonic coupling between low-frequency and high-frequency modes of CO(ads) by using a short-time Fourier transform (STFT) approach to provide an insight into the reaction dynamics for the vertical transfer of CO(ads) adsorbed on the Ag(110) surface.
The ground state properties for CO(ads)–Ag(110) and Ag5(tip)–CO(ads)–Ag(110)
To calculate both electronic properties and STM images, we employed the fully ab initio density functional theory (DFT) code SIESTA.31 The exchange-correlation energy has been considered within the van-der Waal's density functional (vdW-DF)32 to take into account the weak interaction between the CO and Ag(110) surface. The double zeta polarized (DZP) basis set is chosen for all atoms (Ag, C, and O). The localization radii of basis functions are determined from an energy cutoff of 0.01 Ry. The Kohn–Sham orbitals are expanded in a localized basis with a mesh cutoff of 150 Ry. For all adsorbed systems, a periodic 4-atom-layer slab with 15 Å of vacuum and a 2 × 2 × 1 Monkhorst-Pack k-point mesh are used.33 Our calculations of tunneling conductance to generate the STM images are based on the Bardeen approximation as following:| |
 | (1) |
here f(ε) is the Fermi-Dirac function, the energies εt and εs are referred to the Fermi levels of a tip and sample, respectively, and V is the applied bias voltage. The Bardeen's tunneling matrix element (Mts), which couples between sample wavefunction (ψs) and tip wavefunction (ψt) can be represented by a flux through a separation surface (S) as following:| |
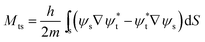 | (2) |
A simple model to evaluate the Bardeen's tunneling matrix is based on the TH approximation in which the wavefunction of a tip (ψt) is approximated as a symmetric s-wave orbital. To include the effect of a tip on adsorption dynamics of adsorbed systems during tunneling-current-induced process, we introduce a nano Ag5 cluster tip in connection with remaining original sample wavefunction (ψs) in our calculations. As a result the Bardeen's tunneling matrix can be derived to be only proportional to LDOS of sample at near Fermi energy as following:
| | |
|Mts|2 ∝ ∑|ψs(x, y, z)|2δ(E − EF) = LDOS(x, y, z, EF)
| (3) |
As temperature is lowered under room temperature, the Fermi-Dirac distribution can be approximated as a step function. Therefore, the eqn (1) used to generate the tunneling conductance can be simply rewritten as following:
| |
 | (4) |
Consequently, the tunneling conductance at a specific position (x,y) can be generated to produce the STM contour maps in a constant-height mode (the height of STM-tip: r0) by integrating the LDOS in 3-dimension spatial grids over a small energy range (δε) at Fermi level (EF). The final form of the total tunneling conductance from a tip into the sample can be derived from the eqn (4) to represent approximately as the LDOS in an x–y plane (Nx and Ny are the total number of grids in the a and b lattice direction).
| |
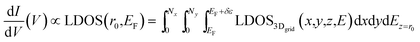 | (5) |
In this study we introduce two kinds of nano Ag5 cluster tips, which is a compressed trigonal-bipyramid tip (Ag5(trigonal-tip)) and a w-shaped planar tip (Ag5(planar-tip)), respectively (as suggested by previous theoretical studies34,35) to investigate the effect of a tip on the adsorption geometries and their tunneling currents for CO(ads) adsorbed on Ag(110) surface. A nano Ag5 cluster tip is placed above the CO(ads)–Ag(110) (the tip-O distance is approximately 3.0 Å). The optimized structures for each adsorbed systems and their corresponding STM images are shown in Fig. 1. Based on the calculated results the average distance between the CO(ads) and Ag(110) become slightly longer (from 2.311 Å to 2.361 Å) as a nano Ag5 cluster tip is introduced and different electronic structure can be observed on the basis of the local density distribution around the Fermi energy, resulting in different STM topographic images. The experimental STM imaging map is also shown in Fig. 1(e) for comparison. To realize the structural effect of the tip on their STM images, we further examine how the frontier molecular electronic states couple to the metallic electronic states with and without the effect of a tip. By analyzing the projected density of states (PDOS) for the CO(ads) molecule and their neighboring Ag surface atom and nano Ag5 cluster tip atoms as shown in Fig. 2, there is a higher intensity of a nano Ag5 cluster tip state around the Fermi energy for the Ag5(planar-tip)–CO(ads)–Ag(110). It is proposed that there is a significantly orbital overlap between a nano Ag5 cluster tip and the surface leading to their strong coupling when the tunneling process occurs. Obviously, the calculated STM image for Ag5(planar-tip)–CO(ads)–Ag(110) can be divided into two groups, the larger bright spot corresponding to the contribution of a nano Ag5 cluster tip state and the central small bright spot as shown in Fig. 1(d) corresponding to that of CO(ads) which are in a good agreement with the experimental STM results.
 |
| | Fig. 1 (a) The optimized structure for CO(ads)–Ag(110), Ag5(trigonal-tip)–CO(ads)–Ag(110), and Ag5(planar-tip)–CO(ads)–Ag(110) and corresponding electronic structures of the LDOS at the Fermi energy. (b) The simulated STM image of CO(ads)–Ag(110), (c) Ag5(trigonal-tip)–CO(ads)–Ag(110), (d) Ag5(planar-tip)–CO(ads)–Ag(110), and (e) the experimental STM result. | |
 |
| | Fig. 2 DOS projected onto the CO molecule (green line) and their neighboring Ag surface atom (blue line) and nano cluster Ag5 tip atom (red line). From the top to bottom: the PDOS of CO(ads) adsorbed on Ag(110), Ag5(trigonal-tip)–CO(ads) adsorbed on Ag(110) and Ag5(planar-tip)–CO(ads) adsorbed on Ag(110). | |
Temperature effects on adsorption dynamics for both CO(ads)–Ag(110) and Ag5(tip)–CO(ads)–Ag(110)
To further investigate the thermal effect as the electron tunneling from a tip into the adsorbed system, we employed the density functional theory-based molecular dynamic (DFT-based MD) simulation, to calculate the CO(ads) adsorbed on the Ag(110) surface with and without a nano Ag5 cluster tip at finite temperature. The DFT-based MD simulations are performed by using the same program SIESTA. All of the model systems are calculated at constant temperature (NVT) using Nosé–Hoover thermostat36,37 for 7.0 ps with a time step of 0.5 fs at 50 K. Based on the experimental conditions, the STM is located above the substrate around the 3.0 Å. Therefore, the distance between the bottom most Ag atom and O atom of CO adsorbed molecule is set to 3.0 Å. After the geometries are optimized, the local minimum structures are used as the initial structural models to run the MD simulation as shown in Fig. 1. During the MD simulation, only the bottom 2 layers of Ag slab and top most Ag atom of a nano Ag5 cluster tip are constrained. The first part of DFT-based MD simulation is to reach an equilibration phase for at least 1.0 ps. Then, the second part of the simulation, which lasts for 6 ps, is followed by collecting data over the temperature-controlled dynamic trajectories to generate a time-dependent autocorrelation function of the derivative of local density of states (ACF − δLDOS(r0,EF)).
The temperature-controlled dynamic trajectories of CO(ads) during 6 ps simulations for each adsorbed-surface systems are examined in Fig. 3(a). First, the adsorbed CO(ads) was found to be stably located on the top site without a tip. Then, as a nano Ag5(trigonal-tip) cluster tip is introduced, the CO(ads) is slightly diffuse above the top site and the flipping rate for CO(ads) is significantly increased. It suggested that the interaction between the CO(ads) and surface is weakening. Finally, when a nano Ag5(planar-tip) cluster tip is introduced, the CO(ads) firstly adsorbed on the top site and then a 180° rotation of CO(ads) occurs at approximately 3 ps to move from the surface to a nano Ag5(planer-tip) cluster tip. To further investigate the electronic properties during the reaction, we collected the trajectories of LDOS at the tip position for the adsorbed systems with and without a tip for each MD simulations. As shown in Fig. 3(b) the tunneling conductance is strongly dependent on the effect of a nano Ag5 cluster tip. The average tunneling conductance of Ag5(planar-tip)–CO(ads)–Ag(110) is found to be more conductive, which is 1.74 and 13.97 times of Ag5(trigonal-tip)–CO(ads)–Ag(110) and CO(ads)–Ag(110), respectively. This tip-enhanced tunneling conductance might increase the coupling between the surface and adsorbate leading to a higher possibility for the transfer process.
 |
| | Fig. 3 (a) The dynamic trajectories of CO(ads) during 6 ps DFTMD simulations and (b) corresponding LDOS(r0,EF) for each investigated systems, CO(ads) adsorbed on Ag(110) (blue line), Ag5(trigonal-tip)–CO(ads) adsorbed on Ag(110) (red line) and Ag5(planar-tip)–CO(ads) adsorbed on Ag(110) (black line). | |
Simulated IETS for CO(ads)–Ag(110) and Ag5(tip)–CO(ads)–Ag(110)
According to Fermi Golden's rule,38 the adsorption line shape of underlying perturbation is generated by considering light-matter cross section interaction and it can be calculated as a Fourier transform of the autocorrelation function by using molecular dynamic simulation. For example, the infrared adsorption spectrum IIR(ω) and Raman spectrum IRaman(ω) can be simulated by a Fourier transform of the dipole moment (μ) autocorrelation function 〈μ(t)μ(0)〉 and polarizability (α) autocorrelation function 〈α(t)α(0)〉, respectively (as discussed by our previous theoretical studies39–42). Following the same strategy, by considering that (1) the IETS is related to the second derivatives of I (tunneling current) with respect to V and (2) the IETS intensity is proportional to the change in LDOS at specific molecular vibration, Qi,  , the IETS can be generated by using a Fourier transform of the autocorrelation of the derivative of LDOS (δLDOS) as following:
, the IETS can be generated by using a Fourier transform of the autocorrelation of the derivative of LDOS (δLDOS) as following:| |
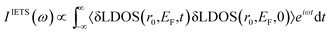 | (6) |
The calculated IETS for CO(ads) adsorbed on the Ag(110) surface for each adsorbed systems are shown in Fig. 4(a). Table 1 lists all the calculated vibrational modes and corresponding experimental IETS results. In the low-frequency region, the IETS active peak at 48 cm−1 (6 meV) can be assigned to the frustrated translation, the active peak at 144 cm−1 and 166 cm−1 (17 meV and 20 meV) can be assigned to doublet frustrated rotation and the active peak at 266 cm−1 (33 meV) is the Ag–C stretching. In the high-frequency region, the IETS active peak at 1991 cm−1 and 2033 cm−1 (246 meV and 252 meV) can be assigned to the C–O stretching. Due to all IETS active vibrational modes are under the low-energy region (<0.5 eV), the Bardeen formalism are still valid. Indeed, the active peaks on IETS by using MD are in a good agreement with the experimental results and even much closer to the experimental results than that of normal mode calculations as shown in Table 1. Furthermore, it is found that the tunneling amplitude of the IETS peaks in low-frequency region is strongly dependent on the influence of a tip. Then, these tip-induced motions in low-frequency region will cause structural changes of CO(ads) adsorbed on the surface. As a result, the lateral hopping and vertical transfer of CO(ads) adsorbed on the Ag(110) surface can be investigated by monitoring the atomic trajectories through molecular dynamics simulations.
 |
| | Fig. 4 The calculated IETS spectrum for each investigated systems, CO(ads) adsorbed on Ag(110) (blue line), Ag5(trigonal-tip)–CO(ads) adsorbed on Ag(110) (red line) and Ag5(planar-tip)–CO(ads) adsorbed on Ag(110) (black line) during 6 ps DFTMD simulations. | |
Table 1 Calculated major IETS active peaks for Ag5(trigonal-tip)–CO(ads) and Ag5(planar-tip)–CO(ads) adsorbed on the Ag(111) surface at 50 K. Normal mode calculation with different type of basis set (SZP, DZ, DZP) and experimental of IETS are also included for comparison
| IETS spectrum |
CO stretching (meV) |
Ag–C stretching (meV) |
Frustrated rotation (meV) |
Frustrated translation (meV) |
| Experimental |
263 |
33 |
18(20) |
5 |
| MD-Ag5-(trigonal-tip) DZP |
255(242) |
34 |
16(20) |
4 |
| Static-Ag5-(trigonal-tip) DZP |
238 |
31 |
16 |
4 |
| Static-Ag5-(trigonal-tip) DZ |
239 |
30 |
15 |
4 |
| Static-Ag5-(trigonal-tip) SZP |
239 |
30 |
15 |
3 |
| MD-Ag5-(planar-tip) DZP |
252(247) |
33 |
18(21) |
6 |
| Static-Ag5-(planar-tip) DZP |
242 |
31 |
14 |
3 |
| Static-Ag5-(planar-tip) DZ |
242 |
30 |
13 |
3 |
| Static-Ag5-(planar-tip) SZP |
242 |
30 |
14 |
2 |
Anharmonic coupling for Ag5(planar-tip)–CO(ads)–Ag(110) by STFT analysis
To further investigate the doublet features which appear in both of the frustrated rotation and C–O stretching, we implement previous Fourier transform of the autocorrelation function of the derivative of LDOS (δLDOS) by introducing the short-time Fourier transform (STFT)41 approach to generate the time-resolved IETS as the following equation:| |
 | (7) |
where h(t + τ) is the rectangular window function, τ is the delay time, IIETS(τ,ω) is a time-resolved power spectrum, which is called a spectrogram of IETS, and T is the window length whose size determines the resolution of frequency of a time-resolved power spectrum. In this study, 2.0 ps of a window length (T) is adopted. As shown in Fig. 5, the spectrogram can provide both of time and frequency domains to explore the evolution of all vibrational modes in IETS for Ag5(planar-tip)–CO(ads)–Ag(110) along the vertical transfer reaction pathway to investigate the correlation between the low-frequency mode and high-frequency modes.
 |
| | Fig. 5 The spectrogram obtained by STFT analysis for the IETS spectrum for the Ag5(planar-tip)–CO(ads) adsorbed on the Ag(110) surface and corresponding structural changed during 6 ps MD simulation. The two kinds of frustrated rotation motion are shown in inserted Figure by SFPF analysis. | |
Based on the structural evolution of CO(ads) adsorbed on Ag(110) surface and corresponding IETS spectrogram the whole vertical transfer process can be divided into three stages. Firstly, before the CO(ads) is transferred to the tip, the frustrated rotation and the C–O stretching are assigned at 166 cm−1 (20 meV) and 1991 cm−1 (246 meV), respectively. Secondly, as the vertical transfer proceeds at nearly 3 ps, the nano Ag5 cluster tip will induce Ag–C bond weakening leading to a red-shifted of the frustrated rotation (from 166 cm−1 to 144 cm−1) and it also accompanies with a blue-shifted of C–O stretching (from 1991 cm−1 to 2033 cm−1). Thirdly, as the CO transfers from the surface to a nano Ag5 cluster tip, the frustrated rotation and C–O stretching are returned to 166 cm−1 (20 meV) and 1991 cm−1 (246 meV), respectively. In contrast to the frustrated rotation, the frustrated translation still remains unchanged (6 meV) along the vertical transfer of CO(ads) on Ag(110) surface. These correlations between low-frequencies (frustrated rotation/frustrated translation) and high-frequency (C–O stretching) imply that there is higher anharmonic coupling for the (frustrated rotation vs. C–O stretching) than that of (frustrated translation vs. C–O stretching). Finally, by using single-frequency pass filter (SPFP)42 analysis scheme, an anharmonic coupling arisen from the frustrated rotation can be further investigated. As shown in inserted figure of Fig. 5, the tip-induced frustrated rotation at 166 cm−1 can be identified to the flipping motion of CO(ads) and it couples to the Ag–C stretching. On the other hand, the tip-induced frustrated rotation at 144 cm−1 can be identified to a rotation along the axis of surface normal and it shows a less dependence on the surface atom due to the weak interaction between the surface and CO(ads) when vertical transfer manipulation occurs.
Conclusion
By combing DFT-based MD simulations with a Fourier transform of autocorrelation function of the derivative of local density of states (FT-ACF-δLDOS), the IETS spectra for CO adsorbed on the Ag(110) surface have been successfully calculated and analyzed to investigate the effect of the nano Ag5 cluster tip on adsorption dynamics and their tunneling conductance at finite temperature during the dynamics process. Our calculated results are summarized below. First, we found that the tunneling conductance generated based on the trajectories of LDOS is significantly increased as a nano Ag5 cluster tip is introduced and their vibrational amplitudes of low-frequency modes (ex. frustrated translation, frustrated rotation and Ag–C stretching) are enhanced in IETS spectrum. Second, the IETS spectrum shows the doublet feature at both low-frequency and high-frequency regions, that is, frustrated rotation and intramolecular C–O stretching, respectively, due to the change of geometry of CO(ads) adsorbed on the Ag(110) surface. Third, by combining the short-time Fourier transform (STFT) and single-frequency pass filter (SPFP) analysis schemes, an anharmonic coupling between the frustrated rotation and intramolecular C–O stretching has been confirmed. Finally, it is our hope that this study will stimulate further experimental efforts to provide more insights into the nature of electron-vibration coupling for investigating the STM-tip induced chemical reactions.
Acknowledgements
The authors would like to thank the National Council in Taiwan for financial support (grant nos. NSC 97-2113-M-032-003-MY3 and NSC 100-2113-M-032-002-) and the National Center for High Performance Computing and Tamkang University in Taiwan for the use of computational facilities.
References
- G. Binnig, H. Rohrer, C. Gerber and E. Weibei, Phys. Rev. Lett., 1982, 49, 57 CrossRef.
- T. A. Jung, R. R. Schlittler and J. K. Gimzewski, Nature, 1997, 386, 696 CrossRef CAS PubMed.
- H. J. Lee and W. Ho, Science, 1999, 286, 1719 CrossRef CAS.
- J. I. Pascual, N. Lorente, Z. Song, H. Conrad and H.-P. Rust, Nature, 2003, 423, 525 CrossRef CAS PubMed.
- L. J. Lauhon and W. Ho, J. Phys. Chem. A, 2000, 104, 2463 CrossRef CAS.
- N. Henningsen, K. Franke, I. Torrente, G. Schulze, B. Priewisch, K. Ruck-Braun, J. Dokic, T. Klamroth, P. Saalfrank and J. Pascual, J. Phys. Chem. C, 2007, 111, 14843 CAS.
- T. Komeda, Y. Kim, Y. Fujita, Y. Sainoo and M. Kawai, J. Chem. Phys., 2004, 120, 5347 CrossRef CAS PubMed.
- D. Riedel, M.-L. Bocquet, H. Lesnard, M. Lastapis, N. Lorente, P. Sonnet and G. Dujardin, J. Am. Chem. Soc., 2009, 131, 7344 CrossRef CAS PubMed.
- J. A. Stroscio and R. J. Celotta, Science, 2004, 306, 242 CrossRef CAS PubMed.
- M. Ohara, Y. Kim and M. Kawai, Jpn. J. Appl. Phys., 2006, 45, 2022 CrossRef CAS.
- B. C. Stipe, M. A. Rezaei and W. Ho, Science, 1998, 280, 1732 CrossRef CAS.
- J. R. Hahn and W. Ho, Phys. Rev. Lett., 2001, 86, 166102 CrossRef.
- T. Komea, Y. Kim, M. Kawai, B. N. J. Persson and H. Ueba, Science, 2002, 295, 2055 CrossRef PubMed.
- N. Mingo and K. Makoshi, Phys. Rev. Lett., 2000, 84, 3694 CrossRef CAS.
- N. Lorente and M. Persson, Faraday Discuss., 2000, 117, 277 RSC.
- N. Lorente and M. Persson, Phys. Rev. Lett., 2000, 85, 2997 CrossRef CAS.
- S. Tikhodeev, M. Natario, K. Makoshi, T. Mii and H. Ueba, Surf. Sci., 2001, 463, 63 CrossRef.
- G. Teobaldi, M. Peñalba, A. Arnau, N. Lorente and W. A. Hofer, Phys. Rev. B: Condens. Matter Mater. Phys., 2007, 76, 235407 CrossRef.
- H. Ren, J. Yang and Y. Luo, J. Chem. Phys., 2009, 130, 134707 CrossRef PubMed.
- J. Tersoff and D. R. Hamann, Phys. Rev. Lett., 1983, 50, 1998 CrossRef CAS.
- J. Tersoff and D. R. Hamann, Phys. Rev. B: Condens. Matter Mater. Phys., 1985, 31, 805 CrossRef CAS.
- F. Demir and G. Kirczenow, J. Chem. Phys., 2012, 136, 014703 CrossRef PubMed.
- X. Li, J. He, J. Hihath, B. Xu, S. M. Lindsay and N. J. Tao, J. Am. Chem. Soc., 2006, 128, 2135 CrossRef CAS PubMed.
- J. Hihath, C. R. Arroyo, G. Rubio-Bollinger, N. Tao and N. Agraït, Nano Lett., 2008, 8, 1673 CrossRef PubMed.
- J. Lykkebo, A. Gagliardi, A. Pecchia and G. C. Solomon, ACS Nano, 2013, 7, 9183 CrossRef CAS PubMed.
- M. Paulsson, T. Frederiksen, H. Ueba, N. Lorente and M. Bradnbyge, Phys. Rev. Lett., 2008, 100, 226604 CrossRef.
- A. Troisi and M. A. Ratner, Nano Lett., 2006, 6, 1784 CrossRef CAS PubMed.
- G. C. Solomon, A. Gagliardi, A. Pecchia, T. Frauenheim, A. D. Carlo, J. R. Reimers and N. S. Hush, J. Chem. Phys., 2006, 124, 094704 CrossRef PubMed.
- M. Paulsson, T. Frederiksen and M. Brandbyge, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 72, 201101(R) CrossRef.
- E. T. R. Rossen, C. F. J. Flipse and J. I. Cerdá, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 235412 CrossRef.
- J. M. Soler, E. Artacho, J. D. Gale, A. García, J. Junquera, P. Ordejón and D. Sánchez-Portal, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 11, 2745 CrossRef.
- G. R-Perez and J. M. Soler, Phys. Rev. Lett., 2009, 103, 096102 CrossRef.
- H. J. Monkhorst and J. D. Pack, Phys. Rev. B: Solid State, 1976, 13, 5188 CrossRef.
- J. Zhao, Y. Luo and G. Wang, Eur. Phys. J. D, 2001, 3, 309 CrossRef.
- B. K. Agrawal, S. Agrawal, P. Srivastava and S. Singh, J. Nanopart. Res., 2004, 6, 363 CrossRef CAS.
- S. Nosé, J. Chem. Phys., 1984, 81, 511 CrossRef PubMed.
- W. G. Hoover, Phys. Rev. A, 1985, 31, 1695 CrossRef.
- D. A. McQuarrie, Statistical Mechanics, Harper and Row, New York, 1976 Search PubMed.
- J. S. Lin, S. Y. Lu, P. J. Tseng and W. C. Chou, J. Comput. Chem., 2012, 33, 1274 CrossRef CAS PubMed.
- S. Y. Lu and J. S. Lin, J. Chem. Phys., 2014, 140, 024705 CrossRef PubMed.
- Y. T. Lee and J. S. Lin, J. Comput. Chem., 2013, 34, 2697 CrossRef CAS PubMed.
- J. P. Su, Y. T. Lee, S. Y. Lu and J. S. Lin, J. Comput. Chem., 2013, 34(32), 2806 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2015 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 and
Jyh-Shing Lin*
and
Jyh-Shing Lin*



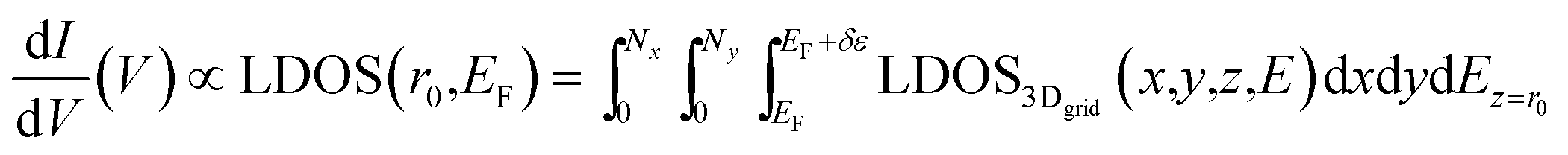



 , the IETS can be generated by using a Fourier transform of the autocorrelation of the derivative of LDOS (δLDOS) as following:
, the IETS can be generated by using a Fourier transform of the autocorrelation of the derivative of LDOS (δLDOS) as following:



