
Isatin N2-diphenylhydrazones: new easily synthesized Vis-Vis molecular photoswitches†
M. Cigáň*,
M. Gáplovský,
K. Jakusová,
J. Donovalová,
M. Horváth,
J. Filo and
A. Gáplovský
Faculty of Natural Sciences, Institute of Chemistry, Comenius University, Mlynská dolina CH-2, SK-842 15 Bratislava, Slovakia. E-mail: cigan@fns.uniba.sk
First published on 16th July 2015
Abstract
The photochromic properties of isatin N2-diphenylhydrazones as a new type of molecular switch were investigated. Interestingly, dimerization stabilizes the formation of only E-isomers in the synthesis, even without a Z-isomer presence in the mother liquor. The irradiation of E-isomers in solution at their absorption maxima leads to the formation of the corresponding Z-isomers. Although the shift in the absorption maximum is not large and photochemical quantum yields are lower compared to in commonly used photoswitches, the absorbance changes are sufficient for potential practical use of these compounds as On-Off switches when the readout wavelength is optimally selected. The process is reversible and switching cycles can be repeated many times in both directions. The determined thermodynamic parameters and calculated geometry of the transition state clearly indicate the inverse mechanism of the thermally initiated slow back Z–E isomerization. Due to the Vis-Vis character of molecular photoswitching and significant geometric changes during the switching process, isatin N2-diphenylhydrazones are eminently suitable for molecular switching in biological/biotechnological applications. Moreover, the interaction of isatin N2-diphenylhydrazones with strongly basic anions increases their functionality and creates a four-state On-Off switch in one molecule. This four-state switch includes the unique E–Z isomerization induced photochemical switching between two stable tautomeric forms of organic amide anions. Simple synthesis of these derivatives and their sufficiently sensitive response to external stimuli promote isatin N2-diphenylhydrazones as suitable candidates for both discussed photochemistry areas (molecular switching and actinometry).
Introduction
The phenomenon of photochromism is defined as a reversible colour change induced in a compound driven in one or both directions by absorption of electromagnetic radiation.1 The back reaction can be driven thermally (photochromism of type T) or photochemically (photochromism of type P). Great strides have been made over the past few decades with commercial UV induced T-type photochromic dyes (spiropyrans, spirooxazines and naphthopyrans) in terms of their colour palette, kinetic control and robustness.2 The largest volume usage of commercial UV induced T-type dyes is in photochromic ophthalmic lens manufacture; especially beneficial for car drivers and motorcyclists.3 Photochromic inks and varnishes for application in a variety of substrates are now a commercial reality.4In addition to aesthetic uses, such as photochromic nail varnish, their colour-change properties have been exploited in more functional applications including security printers which produce optically variable marking to improve document authentication. An important example here is the claim that photochromic material features in US passports.5 Other commercial applications include cosmetics, hair-dye formulations, sunscreen lotions, fishing lines and patented inks for tattoos which are invisible until exposed to sunlight, etc.3
However, because T-type photochromic dyes fade thermally, they are unsuitable for some keenly researched high-tech applications which must rely on P-type photochromism instead.2 Photochromic P-type derivatives have attracted attention because of their huge potential in many practical applications in optics and optoelectronics (optical switches and logic gates, optical memories in data storage, focal plane masks in telescopes, computer generated holograms, optical nanolithography and optical modulation of refractive index in waveguides).6,7 Few novel materials have also been created for information technology by covalently linking P-type dyes to non-photochromic systems in order to photo-regulate properties such as fluorescence.8,9
Widely used classes of photochromic derivatives include diarylethenes, fulgides, spiropyrans, spirooxazines and azobenzenes. While only diarylethenes and furylfulgides are “pure” P-type dyes, relatively simple structural modification can often result in partial T- to P-type dye shift. For example, Temps et al. and Woolley et al. investigated the suitability of azobenzene photoswitching in biological applications.10,11 Most azobenzene-based photoswitches use UV light for photoisomerization. This can limit their application in biological systems, where UV light can trigger unwanted responses, including cellular apoptosis. It is well known that incorporation of ortho or para electron-donating groups in the azo moiety can dramatically red-shift the photoswitching wavelength, but also markedly increases the rate of thermal cis-to-trans relaxation. Short-lived cis isomer means that an intense light source is required in order to maintain substantial fraction of the cis isomer and this presents undesirable limitation in vivo. Woolley et al. found that substitution of all four ortho positions with methoxy groups in an amidoazobenzene derivative leads to substantial red shift of the trans isomer n–π* band, separating it from the cis n–π* transition. This red shift makes trans-to-cis photoswitching possible using 530–560 nm green light. The cis state is thermally stable with a half-life of approximately 2.4 days in aqueous solution in the dark. Reverse (cis-to-trans) photoswitching is accomplished with 460 nm blue light, thus eliminating UV light requirement for bidirectional photoswitching between thermally stable isomers.11 In contrast, Feringa et al. envision the application of Staudinger–Bertozzi azobenzene photoswitches to photocontrol fast biological processes due to the reversible, visible-light-induced switching process and fast thermal relaxation in an aqueous environment.12 Therefore, the development of easily synthesized Vis-Vis (Vis-NIR) T- and P-type photochromic dye remains a distinct challenge in current photochemistry. It should be mentioned here that only hemithioindigos constitute a pure Vis-Vis photoswitch class.
Photochemical reactions responsible for photochromic behaviour of most photoswitches fall into two main categories: isomerization and cyclization. While fulgides, diarylethenes, spiropyrans and dihydroazulenes have been extensively used as molecular photoswitches involving photocyclization and photoreversion processes, five major types of photoswitches based on E–Z isomerization reactions are currently developed. These are azobenzenes, overcrowded alkenes, retinal based switches, hemithioindigos and green fluorescent protein analogues. Excellent reviews are available for both these compound types.13,14
The current trend in the field of photochromism is the structure modification of known type of protochromic derivatives (to simplify the dye preparation, enhance the photochromic response, achieve the accurate dye properties),15–18 new class of photochromic derivatives is rarely discovered.19–21
While photochromism researchers have mainly focused on biological applications over the past two years, a few studies have also examined the technologic application of photochromic switches. Light is an ideal external control for in situ chemical and biological manipulation. A bi-stable molecular photoswitch approach has been used to photoregulate a multitude of important biological processes. These include nucleic acid structure and function, transcription and translation, protein folding, enzyme activity, protein−ligand interactions, peptide structure and function, membrane transport, and receptor modulation and signalling.22,23 Optogenetics has initiated a revolution in neuroscience by enabling simultaneous monitoring and stimulation of specific neuronal populations in intact brain preparations. This is achieved through genetically targeted expression of light sensitive proteins and molecular photoswitches.24,25 Kramer et al. recently introduced a novel strategy for restoring visual function: by adding a synthetic small molecular ‘‘photoswitch’’ which provides light sensitivity to retinal neurons without involving exogenous gene expression.26 Light can also control the formation of vesicles and supramolecular organogels by a cholesterol-bearing amphiphilic molecular switch based on dithienylethene.27 This could provide opportunities in applications as diverse as controlled release of compounds in biological systems, selective labelling and the control of cell growth. Isacoff et al.28 reported a red-shifted, fast-relaxing azobenzene photoswitch for visible-light at 400−520 nm. This controlled an ionotropic glutamate receptor generating a large ionic current on illumination. Further research highlights a responsive, broad-spectrum, ‘smart’ antibiotic temporally activated by light and then auto-inactivated within hours.29 Reversible optical control over active drug concentration provides important pharmacodynamic information. Further, for example, the important reversible noninvasive control over the generation of singlet oxygen has been demonstrated in a bicomponent system comprising a diarylethene photochromic switch and a porphyrin photosensitizer by selective irradiation at distinct wavelengths.30
Additional studies have provided the following technological innovations: a novel dithienylethene-based rewritable hydrogelator with the ability to gelate water has been evolved.31 Shustova et al. published light regulated energy transfer in large light-harvesting using the photoswitch-directed behaviour of metal−porphyrin frameworks.32 Feringa et al. designed UV/vis and NIR light-responsive spiropyran self-assembled monolayers on polycrystalline gold surfaces. Its ultrafast bidirectional switching is attractive for molecular electronics (molecular logic unit and as a memory unit).33 Interestingly, a series of first-generation light-driven molecular motors based on overcrowded alkenes and chiral N-alkyl imines34 has been synthesized.35–40 The combination of ability to rotate repetitively with controlled directionality and its powering by light energy qualifies this system as a rotary molecular motor and distinguishes it from many systems based on molecular switches.41 This has resulted in a variety of applications including dynamic control of intermolecular H-stacking of perylenebisimide, controlling intermolecular through-space magnetic interactions, controlling surface wettability and functioning as a molecular “gear box” and photoswitchable chiral organocatalyst.41
An equally important photochromism application is chemical actinometry.42 An actinometer or dosimeter is a chemical system (fluid, gas, solid, or in a microheterogeneous environment) which undergoes a light-induced reaction at a certain wavelength (λ) for which the quantum yield (Φ(λ)), is accurately known. Measuring the reaction rate enables calculation of the absorbed photon flux crucial in quantitative description of any photochemical reaction. Nowadays, only few actinometers are recommended by IUPAC, which cover visible region of electromagnetic radiation. Here, detection of photochemical conversion is easily monitored by change in absorption: azobenzene (in the 230–460 nm region), ferrioxalate (200–500 nm), 5,12-diphenylnaphthacene (405–500 nm), Aberchrome 540 (reversible photocyclization of fulgide photochromic system; 435–535 nm) and Aberchrome 999P (435–640 nm).42 Unfortunately, azobenzene is classified as a potent toxin (T+), and its reversible thermal reaction complicates calculation of the incident photon flux. Similarly 5,12-diphenylnaphthacene falls in the polyaromatic hydrocarbon health-hazard class, and it is not commercially available in our region. Aberchrome 540 and 999P photochromic actinometers are also no longer commercially available and their preparation from commercially available precursor materials requires several synthetic steps. While ferrioxalate is the most widely used actinometer in the 400–600 nm region,43 its extinction coefficient slightly decreases above 425 nm, and therefore a reliable, easily prepared actinometer in the 400–600 nm region with easy spectrophotometric detection of photochemical transformation is a major challenge in current photochemistry.
In this paper, we focused on isatin N2-diphenylhydrazones as molecular switches and building blocks for the creation of photonic gates (Scheme 1). In the last three years, hydrazones have experienced important application in various supramolecular chemistry areas as molecular switches (photochromism type T; until now only simple chromophoric systems such as benzene, naphthalene and indole were used), photo- and thermo-sensitive supramolecular arrangements and as colorimetric or fluorescent chemosensors.20,44–47 The presence of isatin in proposed new derivatives shift their absorption to the vis-region of the electromagnetic spectrum and isomerization around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N double bond in isatin N2-diphenylhydrazones engenders simple On-Off photochromic ability. Moreover, possible lactam–lactim isatin tautomeric equilibrium enables greater switch function.
N double bond in isatin N2-diphenylhydrazones engenders simple On-Off photochromic ability. Moreover, possible lactam–lactim isatin tautomeric equilibrium enables greater switch function.
 | ||
| Scheme 1 Molecular structure of studied isatin N2-diphenylhydrazones 1 and 2. | ||
The aim of this project therefore was the synthesis and subsequent in-depth research into the photochemical (photochromic) characteristics of the easily synthesized isatin N2-diphenylhydrazones 1 and 2, and their provision of a basic skeleton for a new type of Vis-Vis molecular switch.
Results and discussion
Synthesis
Although photochemical isomerization around the CN double bond has been studied intensively in detail,48,49 it still attracts scientific attention because of the easy and reliable application of light as a stimulus in organic electronics (molecular switches).20,44 The resultant E/Z isomer ratio in the reaction mixture depends particularly on the thermodynamic stability of both isomers. Either the mixture of both E- and Z-isomers or the more energetically favourable isomer is obtained in this synthesis, and the second isomer is generally easily prepared by photochemical conversion.
Surprisingly, in the synthesis of 1 and 2 only E-isomers were isolated from the reaction mixture (Scheme 1). However, theoretical DFT calculations showed that E-isomers E1 and E2 have almost the same stability as the corresponding Z-isomers Z1 and Z2 (Table 1). Although additional solvent (DMF) inclusion slightly favours the E-isomers formation (Table 1), we assume that the intermolecular interactions between the reactants (inducing mutual arrangement of the reactants ensuring the preference to the formation of one isomer in the transition state) and subsequent E-isomer precipitation explain sole E-isomer formation. Similar behaviour with the same conclusion was previously found for isatin N2-phenylsemicarbazones.50 Moreover, mass spectra indicate the presence of E2 dimers in the polar protic solvent (ESI Fig. S1a–c†), therefore the dimerization can additionally stabilize the formation of only one isomer. The Z1 or Z2 formation in mother liquor was not indicated (using 1H NMR spectroscopy). The unexpected low chemical shift of one of the isatin protons in DMF-d7 and benzene-d6 for both E-isomers indicate its shielding from phenyl rings (approximately 5.5 ppm; ESI Fig. S2 and S3†). Selective irradiation of this proton highlighted its spatial interaction with protons in the phenyl ring ortho positions (ESI Fig. S4†). We presume that the phenyl rings in both E-isomers are in equilibrium above the isatin ring, because no chemical shift deviation for particular phenyls was apparent in proton NMR spectra.
| ΔG kJ mol−1 | ||
|---|---|---|
| Vacuum | DMF | |
| E1 | 2 | 0 |
| Z1 | 0 | 7 |
| E1Z1TS | 89 | 96 |
| E2 | 3 | 0 |
| Z2 | 0 | 6 |
| E2Z2TS | 89 | 96 |
UV-vis changes on stimulus application
 | ||
| Fig. 1 UV-vis spectral changes during the isatin N2-diphenylhydrazone E1 solution irradiation at 405 nm and subsequently at 465 nm in DMF (initial E1 concentration cE1 = 1 × 10−4 M; 1 cm cuvette; T = 298.15 K). | ||
The E1 UV-vis spectrum in MeOH also varies with hydrazone concentration (ESI Fig. S6a†). The intensity of the short-wavelength band at 328 nm markedly increases with decreasing hydrazone E1 concentration and the spectrum becomes more diffuse in this region.
The observed hydrazone E1 UV-vis spectrum dependence on concentration results from the presence of intermolecular interactions. The molecules of E1 may interact in solution by hydrogen bonding, forming dimers or higher aggregates (n-mers) at higher concentrations (ESI Scheme S1†). The decrease in hydrazone E1 concentration shifts the equilibrium to the monomeric form accompanied by decreased absorbance at 328 nm. This explanation is consistent with the concentration less-dependent UV-vis spectrum of N-methylated hydrazone E2, with its inability to form inter-monomer hydrogen bonding (ESI Fig. S6b†). However, E2 mass spectra indicate that alternative aggregate types exist in methanolic solution (probably related with π–π interactions).
The reversible molecular configuration change resulting from photochemical and thermally stimulated E–Z CN bond isomerization is a characteristic feature of isatin N2-diphenylhydrazones 1 and 2 (Table 2). The irradiation of E1 solution at 405 nm leads to slight absorbance decrease at the initial long-wavelength absorption maximum and the absorption band shifts bathochromically to longer wavelength (∼18 nm; Fig. 1). A photostationary state is attained after 10 minute irradiation, with no absorption spectral change on further irradiation (ESI Fig. S2†). Compared to commonly used Vis-Vis photoswitches (Vis-Vis azobenzenes, Dronpa and rhodopsine-like chromophores and hemithioindigos), isatin diphenylhydrazones exhibit 10–100 times lower photoisomerization quantum yields, with approximately 60% conversion in the photostationary state (Table 2).1,10,11,14 This will hamper some biological/in-tissue applications. On the other side, the low quantum yield supports their photostability in ambient light (room lights) without the undesired photoswitching interference. Moreover, the extinction coefficients of isatin diphenylhydrazones in the Vis-region are approximately 3–20 times higher than the current azobenzene Vis-Vis switches.10,11 Although the λA shift is not large, absorbance changes are sufficient for potential practical use of these compounds as On-Off switches when the readout wavelength is optimally selected (0.41 a.u. at 465 nm). Very similar photochemical behaviour was noted in all utilized solvents (DMF, MeOH, benzene). Although the λA bathochromical shift apparent in slightly thermodynamically less favourable Z1 and Z2 isomers is quite atypical, the calculated HOMO–LUMO energy gap difference between E- and Z-isomers supports our experimental observations (Table 3).
| Compd | ΦE–Z × 10−3 | t1/2 E–Z s (h) | γE–Z (%) | ΦZ–E × 10−3 | t1/2 Z–E s (h) |
|---|---|---|---|---|---|
| a ΦE–Z – quantum yield for photochemically initiated E–Z isomerization around CN double bond; t1/2 E–Z – half-life related to photochemical E–Z conversion from pure E-isomer to a photostationary state (overall incident photon flux at 405 nm I0 = 5.6 ± 0.1 × 10−4 mol s−1 dm−3); γE–Z – overall photochemical E–Z conversion in a photostationary state (irradiation at 405 nm); ΦZ–E – quantum yield for photochemically initiated back Z–E isomerization around CN double bond; t1/2 Z–E – half-life related to the attainment of a new photostationary state during solution irradiation with light of 465 nm wavelength (back photochemical Z–E conversion from the photostationary state attained at irradiation with light of 405 nm wavelength; overall incident photon flux at 465 nm I0 = 7.7 ± 0.1 × 10−4 mol s−1 dm−3). |
|||||
| 1 | 3.2 ± 0.3 | 22 (0.006) | 61 | 2.6 ± 0.2 | 170 (0.047) |
| 2 | 1.9 ± 0.2 | 29 (0.008) | 60 | 1.4 ± 0.2 | 201 (0.056) |
| HOMO kJ mol−1 | LUMO kJ mol−1 | ΔEHOMO–LUMO kJ mol−1 | Relative ΔEHOMO–LUMO kJ mol−1 | |
|---|---|---|---|---|
| E1 | −678 | −144 | 533 | 18 |
| Z1 | −669 | −154 | 515 | 0 |
| E2 | −676 | −141 | 535 | 17.5 |
| Z2 | −668 | −150 | 518 | 0 |
Three isosbestic points at 312, 345 and 414 nm are observed in the UV-vis spectra during photochemical transformation. When the photostationary equilibrium mixture formed by E-isomer E1 irradiation at 405 nm is subsequently irradiated by light of lower energy (465 nm), the UV-vis spectrum reverts to the starting isomer's initial state.
This process is reversible and switching cycles can be repeated many times in both directions (Fig. 2).
 | ||
| Fig. 2 The absorbance change at 404 nm and 440 nm during the altered irradiation of isatin N2-diphenylhydrazone 1 DMF solution with light of 405 nm and 465 nm wavelength (cE1-initial = 1 × 10−4 M; 1 cm cuvette; T = 298.15 K). | ||
Almost identical high fatigue-resistant photochemical pathways were noted in N1-methylisatin N2-diphenylhydrazone E2-isomer (ESI Fig. S5†). Based on these findings and results from 1H NMR and FTIR spectroscopy, the Z1 and Z2 Z-isomers were identified as products of photochemical reactions. Both Z1 and Z2 undergo thermally initiated back Z–E isomerization at room temperature. The isomerization mechanism has been studied in detail for imines, and to a lesser extent for hydrazones, and in both cases inversion is considered the common pathway mechanism.52 However, in most cases the studies focus on the rates of the isomerization process, and less attention is given to the contributions of enthalpy and entropy to the activation barrier; this can lead to misleading conclusions, as attested by contradictory reports found in the literature.52
Kinetic measurements of Z1 and Z2 thermal isomerization in solvents of different polarity using HPLC chromatography clearly indicate that changes in rate constants (k) or activation Gibbs energies (ΔG‡) with the solvent polarity are not significant (Table 4). Relatively small negative values of the activation entropy ΔS‡ for Z1 and Z2 Z–E isomerization in both solvents further indicate that initial and transition state (TS) differ only little in molecular order and this support the previous assumption.
| Z1 | Z2 | |||||||
|---|---|---|---|---|---|---|---|---|
| k × 10−5 s−1 | ΔS‡ J mol−1 K−1 | ΔH‡ kJ mol−1 | ΔG‡ kJ mol−1 | k × 10−5 s−1 | ΔS‡ J mol−1 K−1 | ΔH‡ kJ mol−1 | ΔG‡ kJ mol−1 | |
| a k – rate constant of the thermally initiated Z–E isomerization at room temperature; ΔS‡ – activation entropy for thermal Z–E isomerization; ΔH‡ – activation enthalpy for thermal Z–E isomerization; ΔG‡ – activation enthalpy for thermal Z–E isomerization. | ||||||||
| Benzene | 7.3 | −39.8 | 96.0 | 96.6 | 5.0 | −39.8 | 110.1 | 97.5 |
| MeOH | 8.2 | −38.0 | 84.6 | 96.0 | 7.6 | −33.5 | 85.8 | 95.8 |
Therefore, we assume that the Z–E isomerization TS has non-polar character and Z–E isomerization can be described by the inverse mechanism, as indicated by calculated TS geometry (ESI Scheme S2†). The –CN–N– moiety in TS is linear and planar to the isatin ring. The free electron pair in TS has p-orbital character and participates in creation of a weak triple –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N– bond and a double –NN–Ph2 bond. This hybridization leads to the CN and NN bond shortening in the TS. Calculated transition state energies ΔG for mutual E/Z isomer transformations (Table 1) correlate perfectly with experimentally determined activation enthalpies ΔG‡ for thermal Z–E isomerization (Table 4). The thermally initiated Z–E isomerization is approximately 20-times slower than the corresponding Z–E photochemical transformation to a photostationary state (t1/2 Z–E thermal = 2.6 h (2.3 h) and 3.9 h (2.5 h) for Z1 and Z2 in benzene (MeOH), respectively).
N– bond and a double –NN–Ph2 bond. This hybridization leads to the CN and NN bond shortening in the TS. Calculated transition state energies ΔG for mutual E/Z isomer transformations (Table 1) correlate perfectly with experimentally determined activation enthalpies ΔG‡ for thermal Z–E isomerization (Table 4). The thermally initiated Z–E isomerization is approximately 20-times slower than the corresponding Z–E photochemical transformation to a photostationary state (t1/2 Z–E thermal = 2.6 h (2.3 h) and 3.9 h (2.5 h) for Z1 and Z2 in benzene (MeOH), respectively).
In contrast to Z1 and Z2 Z-isomers, initial E1 and E2 isomers do not undergo thermal E–Z isomerization in MeOH at room temperature. However, in benzene, both E-isomers isomerize to the corresponding Z-isomers already at room temperature, with rate constants very similar to back Z–E isomerization (although the overall E–Z conversion is smaller than the Z–E back conversion from photostationary state). As previously mentioned, we assume that aggregate formation and its specific stabilization by solvation are responsible for both this effects (isomerization absence in MeOH at room temperature and the overall thermal conversion difference between E–Z and Z–E isomerization). Only increased temperature enables thermal E–Z isomerization in MeOH.
The effect of temperature increase is more significant in E2 methanol solution; most likely due to possible E1 enolization and the corresponding E1-enol higher TS energy (ESI Fig. S7; Scheme S2†).
While the isatin methyl group has little effect on the ΔG‡ value, isatin NH-hydrogen presence is essential for multifunctional switching.
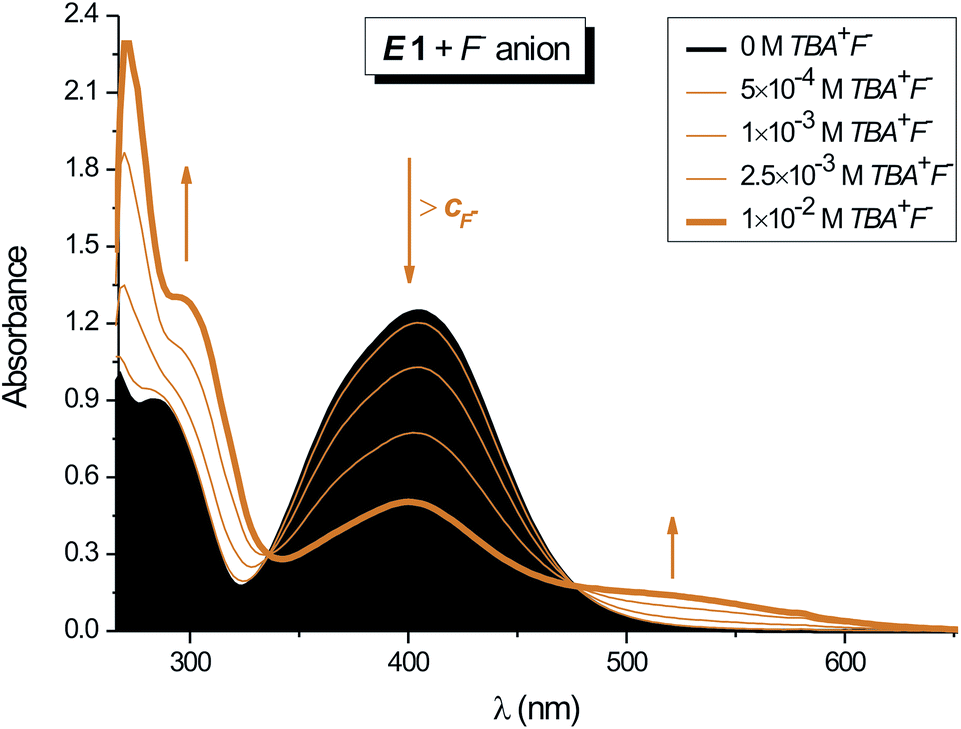 | ||
| Fig. 3 Effect of F− anion concentration on the UV-vis spectrum of isatin N2-diphenylhydrazone E-isomer E1 in DMF (cE1 = 1 × 10−4 M; 1 cm cuvette; T = 298.15 K). | ||
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :60) with strongly basic F− anion in DMF at 298.16 K (determined from absorption spectra)
:60) with strongly basic F− anion in DMF at 298.16 K (determined from absorption spectra)
The isatin N2-diphenylhydrazone E1/Z1 photostationary state mixture has almost the same sensitivity to F− anion as the corresponding E-isomer 1 (Table 5; ESI Fig. S9† enolate formation). The E1/Z1 mixture UV-vis spectrum does not change up to 1 equivalent of anion.
Additional increase in TBA+F− concentration results in slight decrease of absorption band at 400 nm and simultaneous increase in absorption at 300 nm (ESI Fig. S9† enolate formation). The absorption maximum at 420 nm completely disappears at 10−2 M F− concentration and two new absorption bands at 365 and 440 nm are visible in the UV-vis spectrum.
Back F− concentration decrease below 10−3 M (on addition of Z1) leads to backward 420 nm absorption band appearance; thus highlighting the reversibility of this process. The observed different F− concentration effect on the UV-vis spectrum of E1 compared to E1/Z1 mixture can be explained by different steric shield of isatin interaction positions caused by unequal phenyl group location. The isomer conformation affects TBA+F− access to interaction centres in both isomers and thus modifies the structure of the hydrazone/TBA+F− complex. Increased TBA+F− salt concentration leads to gradual NH group deprotonation in E1 (via HF2− formation) and thence to isatin enolate formation (Scheme 2).
 | ||
| Scheme 2 Structure of compound 3 formed from E-isomer E1 at high F− anion excess. | ||
However, large TBA+ cation volume and Z1 spatial configuration prevent enolate formation, and therefore the NH group deprotonation leads only to resonance structure 4 (Scheme 3).
 | ||
| Scheme 3 Structure of compound 4 formed from Z-isomer Z1 at high F− anion excess. | ||
A significant part of the negative charge in structure 4 is localized on the isatin nitrogen, rather than on the oxygen as occurs in structure 3 (see also ESI Table S1 and Scheme S3† structure of compound 4). Despite the small difference in isatin skeleton between parent isomers and products 3 and 4, their room temperature thermal stability indicates the presence of high energy barrier preventing their isomerization around the CN double bond.
Similar to the occurrence in the methylated E2 isomer, F− anion presence does not affect E2/Z2 photostationary state mixture UV-vis spectra.
The OH− anion has almost the same influence on the E1 UV-vis spectrum of as F− anion. Unmethylated Z1 is less sensitive, so that two clearly separated absorption bands over 350 nm appear in the E1/Z1 photostationary state mixture only when OH− concentration is twice that of F− (ESI Fig. S10†).
Remaining anions, including Cl−, Br−, NO2−, NO3−, CH3COO−, ClO4−, SO42−, HSO4− and H2PO4−, do not influence E1 and E1/Z1 absorption spectra shape or intensity because of low isatin NH hydrogen acidity. Hence, only strongly basic anions interact sufficiently strongly with isatin NH hydrogen; as reflected in the UV-vis spectra.
N double bond. Subsequent irradiation of form 4 at its long-wavelength λA with 465 nm light initiates the back photochemical reaction to form 3 (ESI Fig. S11†). UV-vis and 1H NMR spectra of the irradiated reaction mixture are identical with the corresponding spectra of form 3 prepared by mixing isatin N2-diphenylhydrazone E1/Z1 photostationary state mixture with 10−2 M TBA+F− or TBA+OH−. As previously mentioned, the TBA+F− or TBA+OH− interaction with Z1 is limited by hindered TBA+ cation access to the isatin carbonyl oxygen. Photochemical CN isomerization creates the new spatial configuration that eliminates this hindered access. TBA+ cation acts as the counterion to the second tautomeric form of isatin amide anion in this spatial rearrangement (Scheme 4). The diphenylhydrazone conformation change in 3 induced by light thus leads to charge transfer from one tautomeric form of organic amide anion to another.
 | ||
| Fig. 4 Dependence of the UV-vis spectra of hydrazone 3 on the irradiation time in DMF (λexc = 405 nm; cE1 = 1 × 10−4 M; cF− = 1 × 10−2 M; 1 cm cuvette; T = 298.15 K). | ||
 | ||
| Scheme 4 E–Z isomerization induced photochemical switching between two stable tautomeric forms of organic amide anion. | ||
This is unique, in the literature undescribed new type of molecular photoswitching between two thermally stable states of inorganic anion–organic cation complex (at room temperature). Alternate reaction mixture irradiation with light of 405 and 465 nm wavelength enables switch between two 3 and 4 states without observed photodegradation of these states (the presented photochromic system has high fatigue resistance). The switching response is twice as long in the presence of OH− than in F− at the same light intensity.
Although low F− or OH− concentration does not affect the UV-vis spectra of 1 and 2, it exerts observable influence on the photochemical E–Z isomerization equilibrium. After back E–Z isomerization, the E-isomer photostationary concentration in the presence of low F− or OH− concentration is lower than initial E-isomer concentration. Similar to 1 or 2 photoisomerization without anion addition, switching between two stable states without secondary photochemical reaction is also possible in the presence of low F− or OH− concentration. Again, the weakly basic Cl−, Br−, NO2−, NO3−, CH3COO−, ClO4−, SO42−, HSO4− and H2PO4− anions do not influence the E1, E2, Z1 and Z2 UV-vis spectra (in the 0–10−2 M concentration range) and they have the same effect on 1 or 2 photochemical E–Z isomerization as the addition of low F− or OH− concentration.
Conclusion
This paper examined the photochemical (photochromic) properties of two easily synthesized isatin N2-diphenylhydrazones 1 and 2 that could represent the basic skeleton for a new type of Vis-Vis molecular switch. The reversible molecular configuration change resulting from photochemically and thermally stimulated E–Z isomerization of the CN bond is a characteristic feature of the studied isatin N2-diphenylhydrazones (Scheme 5). Although the corresponding photochemical quantum yields of isatin diphenylhydrazones are lower than commonly used photoswitches, the major benefits of this novel switch class are their easy synthesis, easy handling (insensitivity to room lights), their Vis-Vis photoswitching character, marked geometry change during the photoswitching cycle and possible simple additional structure modification.
 | ||
| Scheme 5 Mutual photochemical transformation of the studied E- and Z-isomers. | ||
Moreover, the addition of strongly basic F− or OH− anion to unmethylated E1-isomer solution leads to isatin NH group deprotonation (Scheme 6). This interaction enhances primary E1 switch functionality and delivers four-state On-Off switch formation which could be interesting in both biological and technological photoswitch applications.
 | ||
| Scheme 6 Four-state On-Off switch derived from unmethylated E-isomer E1. | ||
Experimental section
Synthesis
Isatin N2-diphenylhydrazones 1 and 2 were prepared using a modified procedure from the literature .53For further data on the characterization of hydrazones 1 and 2, see ESI† synthesis.
Spectroscopic measurements
Electronic absorption spectra were obtained on a HP 8452A diode array spectrophotometer (Hewlett Packard, USA). All used solvents were UV-spectroscopy grade (Uvasol®, Merck, Germany). DMF was dried with CaH2 and distilled under reduced pressure. Benzene and MeOH were used without further purification. All photochemical measurements were performed at 25 °C in the dark, with only 405 nm or 465 nm LED diodes Thorlabs as light sources with optical power of P = 6 mW and P = 8.5 mW, respectively.Titration experiments
:1 complex stoichiometry were determined using the well-known relation describing the complex anion concentration:54
 | (1) |
For further data on the association constant for hydrazone:anion 1:1 complexes, see ESI† titration experiments.
Light initiated E–Z and Z–E isomerization
For further data on E–Z and Z–E isomerization quantum yield (ΦE–Z) determination for isatin N2-diphenylhydrazones 1 and 2 in DMF solution, see ESI† light initiated E–Z and Z–E isomerization.
Quantum-chemical calculations
The relative stabilities of isatin-diphenylhydrazone conformers and transition states have been investigated by use of quantum-chemical calculations. The geometries of the structures were optimized at the M062x 6-31+g(dp) level. Stationary points were characterized as minima by computations of harmonic vibrational frequencies at the same theory level as geometry optimization. Single point energies have been further calculated at M062X 6-311++G(dp) level. The zero-point vibrational energies and thermal corrections to the free energies were determined by using the unscaled M062x 6-31 + g(dp) frequencies. Free energies of solvated structures were calculated using IEF-PCM method. All calculations were carried out with the Gaussian 09 program package.56Mass spectroscopy
The mass spectra were analyzed on an Agilent 1200 HPLC system with autosampler with single quadrupole mass spectrometer 6110A (Agilent Technologies, USA). Samples were injected directly without separation to mass spectrometer in methanol and acetonitrile, respectively. The following positive multimode ion source (combination of ESI† and APCI) were utilized. The capillary voltage was 2.0 kV, the source block and drying gas temperatures were 230 °C and 350 °C, respectively. The drying gas (nitrogen) and nebulizing gas (nitrogen) were set at 5.0 L min−1 and 40 psi, respectively. Mass spectra were acquired in mass range from 80 to 800 amu.NMR spectroscopy
All NMR experiments were recorded on Varian VNMRS 600 MHz spectrometer in 5 mm NMR tube.Acknowledgements
This contribution is the result of the projects implementation (ITMS 26240220086 and ITMS 26240220072) supported by the OPRaD funded by the ERDF.Notes and references
- H. Bouas-Laurent and H. Dürr, Pure Appl. Chem., 2001, 73, 639–665, DOI:10.1351/pac200173040639.
- A. Towns, Chem. Ind., 2012, 76, 32–35, DOI:10.1002/cind.7605_11.
- S. N. Corns, S. M. Partington and A. D. Towns, Color. Technol., 2009, 125, 249–261, DOI:10.1111/j.1478-4408.2009.00204.x.
- SolarActive International Inc, (available at http://solaractiveintl.com, last accessed, November 2014).
- S. Higgins, Chem. Br., 2009, 39, 26–29 Search PubMed.
- C. Bertarelli, A. Bianco, R. Castagna and G. Pariani, J. Photochem. Photobiol., C, 2011, 12, 106–125, DOI:10.1016/j.jphotochemrev.2011.05.003.
- C. Yun, J. You, J. Kim, J. Huh and E. Kim, J. Photochem. Photobiol., C, 2009, 10, 111–129, DOI:10.1016/j.jphotochemrev.2009.05.002.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327–336, 10.1039/B400387J.
- I. Yildiz, E. Deniz and F. M. Raymo, Chem. Soc. Rev., 2009, 38, 1859–1867, 10.1039/B804151M.
- R. Siewertsen, H. Neumann, B. Buchheim-Stehn, R. Herges, Ch. Näther, F. Renth and F. Temps, J. Am. Chem. Soc., 2009, 131, 15594–15595, DOI:10.1021/ja906547d.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687, DOI:10.1021/ja209239m.
- C. Poloni, W. Szymański, L. Hou, W. R. Browne and B. L. Feringa, Chem.–Eur. J., 2014, 20, 946–951, DOI:10.1002/chem.201304129.
- B. L. Feringa and W. R. Browne, Mol. Switches, 2011 DOI:10.1002/9783527634408.
- C. García-Iriepa, M. Marazzi, L. M. Frutos and D. Sampedro, RSC Adv., 2013, 3, 6241–6266, 10.1039/C2RA22363E.
- H. H. Liu and Y. Chen, J. Phys. Chem. A, 2009, 113, 5550–5553, DOI:10.1021/jp810919j.
- Z. Li, Y. Lin, J. L. Xia, H. Zhang, F. Fan, Q. Zeng, D. Feng, J. Yin and S. H. Liu, Dyes Pigm., 2011, 90, 290–296, DOI:10.1016/j.dyepig.2010.09.015.
- T. Kudernac, T. Kobayashi, A. Uyama, K. Uchida, Sh. Nakamura and B. L. Feringa, J. Phys. Chem. A, 2013, 117, 8222–8229, DOI:10.1021/jp404924q.
- Ch. Böttcher, G. Zeyat, S. A. Ahmed, E. Irran, T. Cordes, C. Elsner, W. Zinth and K. Rueck-Braun, Beilstein J. Org. Chem., 2009, 5, 1–7, DOI:10.3762/bjoc.5.25.
- L. Liu, H. Sun, S. Abdurehman, D. Jia, J. Guo and D. Wu, J. Photochem. Photobiol., A, 2013, 267, 106–125, DOI:10.1016/j.jphotochem.2013.06.021.
- M. N. Chaur, D. Collado and J. M. Lehn, Chem.–Eur. J., 2011, 17, 248–258, DOI:10.1002/chem.201002308.
- S. Helmy, F. A. Leibfarth, S. Oh, J. E. Poelma, C. J. Hawker and J. Read de Alaniz, J. Am. Chem. Soc., 2014, 136, 8169–8172, DOI:10.1021/ja503016b.
- W. Szymański, J. M. Beierle, H. A. V. Kistemaker, W. A. Velema and B. L. Feringa, Chem. Rev., 2013, 113, 6114–6178, DOI:10.1021/cr300179f.
- W. A. Velema, M. Van der Toorn, W. Szymański and B. L. Feringa, J. Med. Chem., 2013, 56, 4456–4464, DOI:10.1021/jm400115k.
- E. Papagiakoumou, Biol. Cell, 2013, 105, 1–22, DOI:10.1111/boc.201200087.
- R. H. Kramer, A. Mourot and H. Adesnik, Nat. Neurosci., 2013, 16, 816–823, DOI:10.1038/nn.3424.
- I. Tochitsky, A. Polosukhina, V. E. Degtyar, N. Gallerani, C. M. Smith, A. Friedman, R. N. Van Gelder, D. Trauner, D. Kaufer and R. H. Kramer, Neuron, 2014, 81, 800–813, DOI:10.1016/j.neuron.2014.01.003.
- J. T. Van Herpt, J. Areephong, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem.–Eur. J., 2014, 20, 1737–1742, DOI:10.1002/chem.201302902.
- M. A. Kienzler, A. Reiner, E. Trautman, S. Yoo, D. Trauner and E. Y. Isacoff, J. Am. Chem. Soc., 2013, 135, 17683–17686, DOI:10.1021/ja408104w.
- A. W. Velema, J. P. Van der Berg, M. J. Hansen, W. Szymański, A. J. M. Driessen and B. L. Feringa, Nat. Chem., 2013, 5, 924–928, DOI:10.1038/NCHEM.1750.
- L. Hou, X. Zhang, T. C. Pijper, W. R. Browne and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 910–913, DOI:10.1021/ja4122473.
- J. T. Van Herpt, M. C. A. Stuart, W. R. Browne and B. L. Feringa, Chem.–Eur. J., 2014, 20, 3077–3083, DOI:10.1002/chem.201304064.
- D. E. Williams, J. A. Rietman, J. M. Maier, R. Tan, A. B. Greytak, M. D. Smith, J. A. Krause and N. B. Shustova, J. Am. Chem. Soc., 2014, 136, 11886–11889, DOI:10.1021/ja505589d.
- O. Ivashenko, J. T. Van Herpt, B. L. Feringa, P. Rudolf and W. R. Browne, Langmuir, 2013, 29, 4290–4297, DOI:10.1021/la400192c.
- L. Greb and J. M. Lehn, J. Am. Chem. Soc., 2014, 136, 13114–13117, DOI:10.1021/ja506034n.
- K. Y. Chen, S. J. Wezenberg, G. T. Carroll, G. London, J. C. M. Kistemaker, T. C. Pijper and B. L. Feringa, J. Org. Chem., 2014, 79, 7032–7040, DOI:10.1021/jo501190f.
- A. Cnossen, J. C. M. Kistemaker, T. Kojima and B. L. Feringa, J. Org. Chem., 2014, 79, 927–935, DOI:10.1021/jo402301j.
- G. London, G. T. Carroll and B. L. Feringa, Org. Biomol. Chem., 2013, 42, 3477–3483, 10.1039/c3ob40276b.
- J. Conyard, A. Cnossen, W. R. Browne, B. L. Feringa and S. R. Meech, J. Am. Chem. Soc., 2014, 136, 9692–9700, DOI:.1021/ja5041368.
- K. Y. Chen, O. Ivashenko, G. T. Carroll, J. Robertus, J. C. M. Kistemaker, G. London, W. R. Browne, P. Rudolf and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 3219–3224, DOI:10.1021/ja412110t.
- J. Bauer, L. Hou, J. C. M. Kistemaker and B. L. Feringa, J. Org. Chem., 2014, 79, 4446–4455, DOI:10.1021/jo500411z.
- J. Chen, J. C. M. Kistemaker, J. Robertus and B. L. Feringa, J. Am. Chem. Soc., 2014, 136, 14924–14932, DOI:10.1021/ja507711h.
- H. J. Kuhn, S. E. Braslavsky and R. Schmidt, Pure Appl. Chem., 2004, 76, 2105–2146, DOI:10.1351/pac200476122105.
- T. Lehóczki, É. Józsa and K. Ősz, J. Photochem. Photobiol., A, 2013, 251, 63–68, DOI:10.1016/j.jphotochem.2012.10.005.
- X. Su and I. Aprahamian, Chem. Soc. Rev., 2014, 43, 1963–1981, 10.1039/c3cs60385g.
- G. Vantomme and J. M. Lehn, Angew. Chem., Int. Ed., 2013, 52, 3940–3943, DOI:10.1002/anie.201210334.
- G. Vantomme and J. M. Lehn, Chem.–Eur. J., 2014, 20, 1–7, DOI:10.1002/chem.201404561.
- G. Vantomme, S. Jiang and J. M. Lehn, J. Am. Chem. Soc., 2014, 136, 9509–9518, DOI:10.1021/ja504813r.
- L. L. Costanzo, U. Chiacchio and S. Giuffrida, J. Photochem., 1980, 14, 125–132, DOI:10.1016/0047-2670(80)80003-7.
- S. Zilberg and Y. Haas, Photochem. Photobiol. Sci., 2003, 2, 1256–1263, 10.1039/B306137J.
- K. Jakusová, M. Gáplovský, J. Donovalová, M. Cigáň, H. Stankovičová, R. Sokolík, J. Gašpar and A. Gáplovský, Chem. Pap., 2013, 67, 117–126, DOI:10.2478/s11696-012-0248-x.
- M. T. Silva and J. C. Netto-Ferreira, J. Photochem. Photobiol., A, 2004, 162, 225–229, DOI:10.1016/S1010-6030(03)00383-6.
- S. M. Landge, E. Tkatchouk, D. Benítez, D. A. Lanfranchi, M. Elhabiri, W. A. Goddard III and I. Aprahamian, J. Am. Chem. Soc., 2011, 133, 9812–9823, DOI:10.1021/ja200699v.
- F. D. Popp, J. Heterocycl. Chem., 1984, 6, 1641–1645, DOI:10.1002/jhet.5570210614.
- B. Valeur, J. Pouget, J. Bourson, M. Kaschke and N. P. Ernsting, J. Phys. Chem., 1992, 96, 6545–6549, DOI:10.1021/j100195a008.
- A. Gáplovský, Š. Toma and J. Donovalová, J. Photochem. Photobiol., A, 2007, 191, 162–166, DOI:10.1016/j.jphotochem.2007.04.018.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, revision A.1, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Including additional data for Experimental section, Schemes S1–S3, Table S1, Fig. S1–S10 and additional References. See DOI: 10.1039/c5ra06625e |
| This journal is © The Royal Society of Chemistry 2015 |