
Metabolic pathways in cancers: key targets and implications in cancer therapy
Abstract
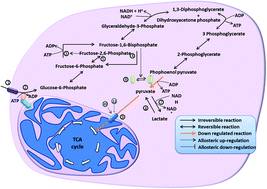
Proliferation and self-sufficiency are two of the most important properties of cancer cells. Although genetic aberrations are believed to be the reason for cancer development, the importance of metabolic alterations in cancer development has been in the lime light lately. The most challenging aspect in cancer treatment has been the similarity to host cells. The discovery of various metabolic alterations that occur in cancers to attain and maintain a proliferative state has resulted in new information on the metabolic differences between normal and cancer cells. One such alteration is the establishment of the Warburg effect. This review elaborates on various changes that lead to the establishment of the Warburg effect in cancer cells and their consequences. Understanding the metabolic uniqueness of various cancers can aid in the identification of novel molecular targets leading to more efficient strategies in cancer treatment.
 Please wait while we load your content...
Please wait while we load your content...

