
Unique lead adsorption behavior of ions sieves in pellet-like reduced graphene oxide†
Dejian Chena,
Shunxing Li*ab,
Liling Zoua and
Fengying Zheng‡
ab
aDepartment of Chemistry and Environmental Science, Minnan Normal University, Zhangzhou, Fujian 363000, China. E-mail: lishunxing@mnnu.edu.cn; shunxing_li@aliyun.com; Fax: +86 596 2591395; Tel: +86 596 2591395
bFujian Province Key Laboratory of Modern Analytical Science and Separations Technology Minnan Normal University, Zhangzhou, Fujian, China
First published on 24th August 2015
Abstract
Hollow and pellet-like reduced graphene oxide (P-RGO) was synthesized by electrostatic assembly and acid-treatment. The average size and shell thickness of P-RGO were 65.9 nm and 4 nm, respectively. The adsorption capacity of P-RGO for lead ions was as high as 184.5 mg g−1 and the adsorption equilibrium of lead ions (50 mg L−1) was achieved within 5 min. Lead ion removal by adsorption onto P-RGO increased with decreasing pH. Because P-RGO exhibited stronger affinity for lead ions than for water, lead ions could be drawn and stored into the cavity in the P-RGO, i.e., ions storage effect was observed. This led to high adsorption capacity and abnormal adsorption behavior. The adsorption mechanism was proposed as ions exchange and competitive process for layer RGO and P-RGO, respectively. This study suggested that structure reconstruction of materials could enhance the efficiency and capacity for unique metal removal.
1 Introduction
The aquatic ecosystems and public health are threatened by lead pollution for its toxicity,1 bioaccumulation,2–4 and non-biodegradation. Thus, effective lead ion removal from water bodies is required. In comparison with current treatment methods for lead contamination, such as ions exchange,5 cloud point extraction,6 coprecipitation,7 membrane filtration,8 and reverse osmosis,9 adsorption is one of the most attractive options due to its good removal performance, operation simplicity, and low cost.10 To improve adsorption performance, two dimensional materials with Pb specificity and high surface area are developed, including graphene nanosheets and titanium carbide.11,12Graphene, a two-dimensional carbon atom layer with sp2 hybridization, is a basic structure unit of zero dimensional fullerene, one dimensional carbon nanotube, and three-dimensional graphite.13 Due to unique electrical properties,14,15 high heat conductivity,16 mechanical strength, and specific surface area,17 graphene-based materials are widely used in analytical science, environmental science, and biological medicine. However, the large surface area and strong π–π interaction between graphene layers lead to facile stacking and resulting limitation on their applications.18 Methods, such as vacuum filter,19,20 chemical vapor depositions,21–23 wet spinning,24 and self-assembly,25–27 have been extensively explored to prepare three-dimensional structure of graphene to prevent such stacking. The 3D graphene, including graphene-based aerosol28–30 and graphene-coated nanoparticles,31–33 exhibits large surface area and high adsorption capacity. However, sparse information on the preparation of uniform pellet-like reduced graphene oxide (P-RGO) is available.34
Graphene-based materials, specifically for graphene oxide, with large surface areas and abundant active sites are used for metal and dye removal.11,35–38 When immersed in water, graphene-based materials can act as molecular sieves.39 However, their applications are limited by the difficulty in their separation from aqueous solution for their high water solubility. The adsorption in nearly neutral aqueous media gives a further challenge to acidic wastewater. Herein, hollow and P-RGO is synthesized by electrostatic assembly and acid-treatment, because P-RGO with multilayer graphene walls and suitable surface functional groups can be used as a sieve for lead removal. The adsorption performance and mechanism of lead ions onto P-RGO are tested by structural characterization and batch adsorption experiment.
2 Experimental sections
2.1 Chemicals and materials
Graphite powder was purchased from national medicine group chemical reagent Co., Ltd in China. Sodium nitrate, potassium permanganate, hydrochloric acid, sulfuric acid, barium chloride and hydrogen peroxide (30%) were analytical grade and purchased from Shantou west long chemical Co., Ltd in China. Iron(III) chloride anhydrous (98%) was purchased from Alfa Aesar. All chemicals and solvents were used as received. All aqueous solution were prepared using ultrapure water (18 MU) from a Milli-Q system (Millipore).2.2 Preparations of graphene oxide
Graphene oxide (GO) was prepared according to modified Hummer method.40 Typically, graphite powder (1.0 g) was mixed with concentrated sulfuric acid (23 mL) in a 1000 mL round bottom flask and stirred in an ice bath. NaNO3 (0.5 g) and KMnO4 (3.0 g) were slowly added into the suspensions and kept at 0 ± 1 °C. After keeping the mixture at 35 ± 3 °C for 30 min, water (46 mL) was slowly added, the suspensions was heated up to 98 °C and maintained for 15 min, and then water (140 mL) and H2O2 (30%, 2 mL) were added to end the reactions. Thereafter, the suspensions were hot filtered, washed by HCl solution (5%) until no SO42− in the filtrate was detected. The products were vacuum-dried at 60 °C overnight and then sealed for the preservation.2.3 Preparations of P-RGO and RGO
P-RGO was prepared by a simple two-step reaction, as shown in Fig. 1. Reduced graphene oxide-coated α-Fe2O3 pellet was prepared as follows. Ferric chloride (1.0 g) and anhydrous sodium acetate (1.0 g) were dissolved in diethylene glycol (20 mL) to form a homogeneous solution and then graphene oxide solution (5.0 mg mL−1, 5 mL) was added. The mixture was dispersed by ultrasound for 30 min, transferred into a Teflon-lined stainless-steel autoclave, heated at 200 °C for 10 h, and cooled to room temperature. The black products were washed several times with ethanol and water. The brownish red solid was dried at 80 °C and treated with HCl (6 mol L−1, 30 mL) for one day. After that, the mixture was filtered with 0.22 μm filter membrane and washed with ultrapure water until the filtrate was neutral. The black and feathery solid was dried at 80 °C and then P-RGO was obtained. The synthesis procedure of RGO was similar to that of P-RGO, except that no ferric chloride was added.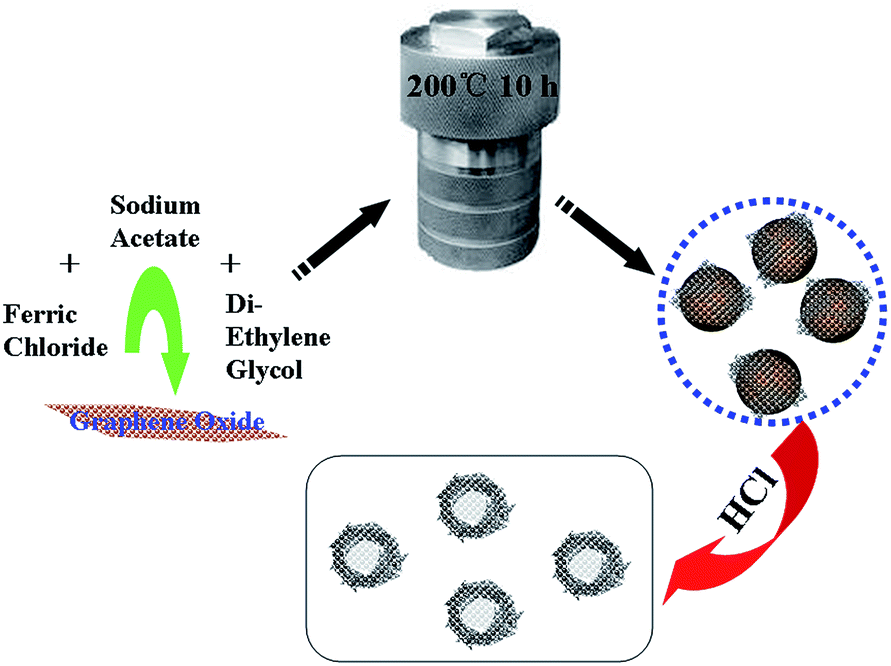 | ||
| Fig. 1 A schematic illustration of the formation process of pellet-like reduced graphene oxide. | ||
2.4 Batch adsorption experiments
At room temperature, a weighed P-RGO (or RGO) was added into a 100 mL beaker containing 25 mL of lead ions solution (pH = 5.6, 10–80 mg L−1) and dispersed by the ultrasound. After adsorption at different time intervals, the above suspension was separated using a vacuum filter with 0.22 μm membrane and the concentration of lead in the filtrate was determined by flame atomic absorption spectrum (FAAS). The adsorption capacity (qe) and adsorption ratio of RGO (or P-RGO) were calculated according to the following equations:
where Ci and Ce are the initial and equilibrium concentrations of lead ions (mg L−1), respectively, V is the volume of solution (mL), and w is the weight of adsorbent (g).

2.5 Desorption experiments
To test the desorption behavior of P-RGO (or RGO) toward lead ions, the lead ions adsorbed on P-RGO (or RGO) were used from batch adsorption experiments. After adsorption equilibrium, the mixture was filtered with 0.22 μm membrane, the black solid was washed several times with ultrapure water and then dried rapidly at 100 °C. The obtained solid was mixed with 25 mL of ultrapure water for different time intervals (0, 10, 30, 60, 120, 240, 360, or 480 min). Lastly, the suspension was separated using a vacuum filter with 0.22 μm membrane, then the concentration of lead ions desorbed from P-RGO (or RGO) into the filtrate (Cds) was determined by FAAS. The desorption efficiency (αds) was obtained according to the following equations:| wad = (Ci − Ce)V |
| wds = CdsV |
where Ci and Ce are the initial and equilibrium concentrations of lead ions (mg L−1), respectively, Cds is the concentrations of lead ions desorbed from P-RGO (or RGO) (mg L−1), V is the volume of solution (mL), wad is the weight of lead ions adsorbed on P-RGO (or RGO) (mg), and wds is the mass of lead ions desorbed in aqueous solution (mg).

2.6 Characterizations methods
The morphology of nanocomposites during preparations process and final products were characterized by scanning electron microscopy (SEM, Hitachi S-4800) with an accelerating voltage of 10 kV. EDX (Horiba EMAX7593-H) was used for element analysis. Using Cu-Kα radiation (λ = 0.1541 nm), the X-ray diffraction (XRD) patterns of all samples were recorded with a D/MAX-TTRIII diffractometer (Rigaku corporation, Japan) and the data were collected from 10° to 70° (2θ). Transmissions electron microscopy (TEM) was obtained with Tecnai G2 20 S-TWIN. X-ray photoelectron spectrum (XPS) was carried out on Horiba EMAX7593-H (Japan). Raman spectrum was performed by a laser Raman spectrometer (Renishaw inVia plus, UK). The concentration of lead ions was measured by FAAS (GBC Co., Australia). The P-RGO was degassed at 300 °C for 12 h under vacuum and then its nitrogen adsorption isotherm at −196.15 °C was obtained using a Quantachrome, Asic-7 physisorption analyzer. The surface area of P-RGO or RGO was evaluated by the Brunauer–Emmett–Teller (BET) model, while the pore size and pore volume were estimated with Barrett–Joyner–Halenda theory.3 Results and discussions
3.1 Morphology and structure of GO, α-Fe2O3, α-Fe2O3@graphene, and P-RGO
The SEM image (Fig. 2a) clearly confirmed the layered structure of synthesized GO with micrometer sizes, indicating the formation of GO sheets through chemical oxidation and ultrasonic exfoliation. Fig. 2b showed the SEM image of α-Fe2O3 prepared without GO, it clearly indicated that the particles were micrometer level, uneven distributions, and serious agglomerations. However, when GO was added into the reaction, the monodispersion, nanosize, and uniform elliptical nanoparticles could be synthesized (Fig. 2c). No large layers of graphene were observed. EDS spectrum (Fig. S1†) displayed electron image of iron, carbon, and oxygen, which demonstrated iron and oxygen were the main components of the composites, while carbon content was less. GO served both as a support for the growth of nanoparticles and a surface modifier. The α-Fe2O3 was removed by the addition of 6 mol L−1 of HCl, which resulted in the formations of P-RGO. The nanoparticles of P-RGO (shown in Fig. 2d), as well as the untreated one (i.e., α-Fe2O3@graphene, Fig. 2c), were monodisperse and elliptical shape indicating the direct attachment of RGO onto the surface of α-Fe2O3 as shell–core structure. Size distribution of hollow graphene spheres (Fig. S2†) showed the P-RGO was uniform with an average size of 65.9 nm. | ||
| Fig. 2 Typical SEM images of GO (a), Fe2O3 NPs without GO (b), Fe2O3@graphene nanoparticles (c) and pellet-like reduced graphene oxide (d). | ||
To further observe the morphology and structure of the composites, TEM was used to characterize the three dimensional structure of α-Fe2O3@graphene composites. As shown in Fig. 3a and b, the composites exhibited a notable shell–core structure and their core and shell were α-Fe2O3 and graphene, respectively. The graphene sheets could be uniformly coated onto α-Fe2O3 particles, yielding elliptical morphology. After dissolving of α-Fe2O3 with HCl (6 mol L−1) for 90 min, a yolk–shell structure was formed and the thickness of outer reduced graphene oxide layer was about 4 nm (shown in Fig. 3c). After removing the α-Fe2O3 by excessive HCl, a hollow pellet like structure with some wrinkles on the surface was formed (Fig. 3d). Acid treatment did not alter the structure of the outer carbon shells, i.e., a kind of pellet like structure.
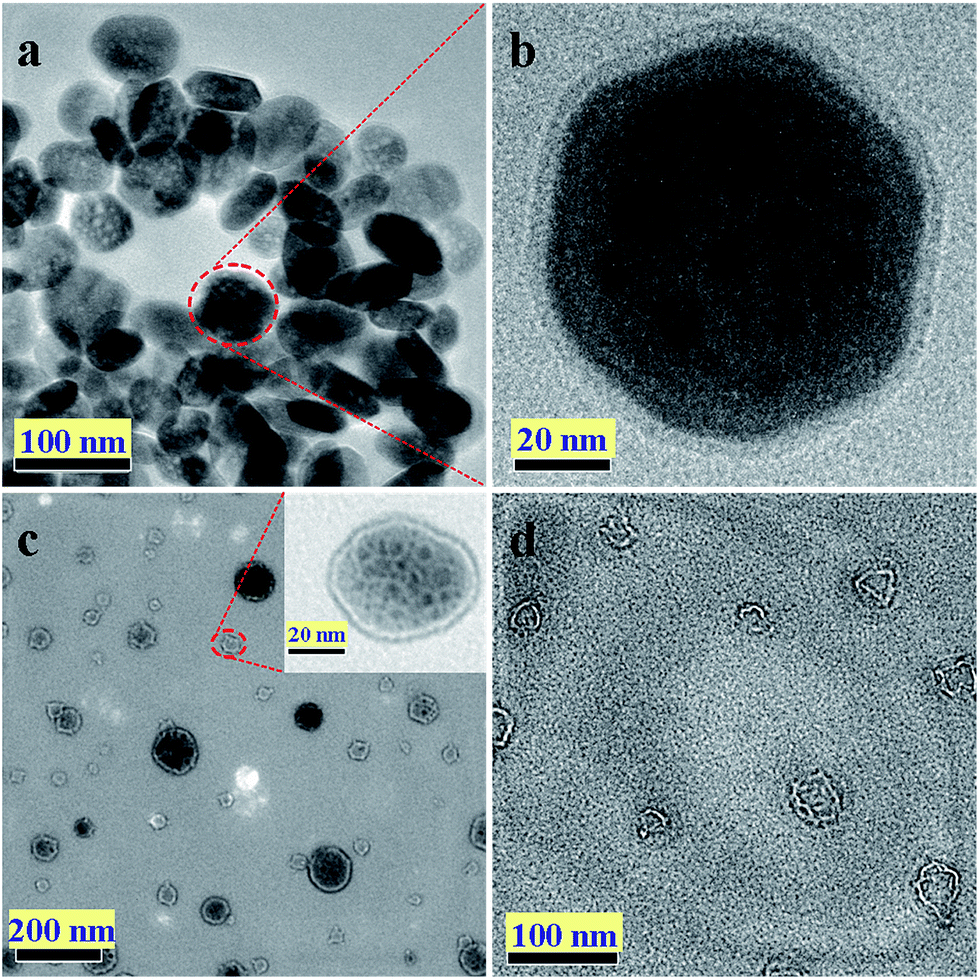 | ||
| Fig. 3 Typical TEM images of Fe2O3@graphene nanoparticles (a) and (b), Fe2O3@graphene nanoparticles after acid treated for 60 min (inset is a single particle) (c) and pellet-like reduced graphene oxide after acid treatment for 10 h (d). | ||
In Fig. 4a, Raman spectra of the P-RGO had two strong peaks in 1604 and 1356 cm−1, corresponding to G and D bands, respectively. The G band was usually assigned to first-order scattering of the E2g mode from the sp2 carbon domains, while the D band was associated with structural defects, amorphous carbon, or the edges that broke the symmetry and selection rule. The ratio of D to G band intensity of our sample was relatively intense compared to the GO, which was in agreement with previous result for graphene sample obtained from GO. This increase might be caused by the defects within the sp2 carbon network that arose upon reduction of the exfoliated GO. Because the synthesized P-RGO was multilayer graphene stack, its weak was peak around 2729 cm−1, which was bigger than that of single-layer graphene (2700 cm−1). This was consistent with the structure of TEM characterizations. A combination band of D and G induced by the disorder at 2930 cm−1 showed that P-RGO contained highly disordered and randomly arranged graphene sheets. The XRD pattern of RGO and P-RGO was showed in Fig. S3.† The weak and broad peak at 11.7° and a shape peak at 25.8° were corresponded to the interlayer distances of 7.6 Å and 3.53 Å, respectively. The XRD peak of RGO and P-RGO was similar and showed the result of removing intercalated water molecules and the oxide groups by hydrothermal method. According to the linear region of the graph in Fig. 4b and the BET equation, the BET specific surface area of the as-prepared P-RGO was 288.85 m² g−1, which was much higher than that of RGO (16.75 m² g−1, Table S1†). The increased specific surface area was attributed to the hollow and pellet like structure of P-RGO.
 | ||
| Fig. 4 Raman spectra (a) and nitrogen adsorption and desorption isotherms (b) of pellet-like reduced graphene oxide. | ||
The chemical states of elements in P-RGO were further investigated by XPS. The XPS survey spectra of the P-RGO (Fig. 5a) showed that P-RGO was mainly composed of C and O, and a small number of Fe and Cl which were residual from α-Fe2O3 and HCl, respectively. Fig. 5b showed the high-resolution spectra of C1s which were isolated into three different peaks. The strong peak at 284.8 eV was corresponded to the C atom in C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond. The banding energy at 286.6 and 287.5 eV were attributed to the C atom in C–O and CO, respectively. From the XPS analysis, it was clearly showed that the GO was highly reduced after hydrothermal reactions but P-RGO still contained a small amount of oxygen groups on the surface which resulted in a certain water solubility and then offered enough affinity for lead ions in water.
C bond. The banding energy at 286.6 and 287.5 eV were attributed to the C atom in C–O and CO, respectively. From the XPS analysis, it was clearly showed that the GO was highly reduced after hydrothermal reactions but P-RGO still contained a small amount of oxygen groups on the surface which resulted in a certain water solubility and then offered enough affinity for lead ions in water.
 | ||
| Fig. 5 X-ray photoelectron spectroscopy spectrum (a) and the corresponding high-resolution spectra of C1s (b) for pellet-like reduced graphene oxide. | ||
3.2 Adsorption performance of RGO and P-RGO toward lead(II) ions
A series of batch adsorption tests for adsorption performances toward lead(II) ions onto RGO and P-RGO were performed using the reported methods.11 The effect of the contact time on adsorption was tested using 50 mg L−1 of lead ions at 25 °C. The pH value 5.6 was selected because it was the pH value of CO2 saturated solution and the critical value for acid rain. Lead ions could be adsorbed quickly both on RGO and P-RGO and its adsorption equilibrium was reached in 5 min (Fig. 6), which was attributed to its structure and specific surface area. Fig. 7a showed the effect of initial concentration on the adsorption capacity of RGO and P-RGO. The adsorption capacity increased as increasing lead ions concentration until the adsorption equilibrium was reached. Under the same conditions, the adsorption capacity of P-RGO was higher than that of RGO. When lead ions concentration was more than 100 mg L−1, the increase rate of lead ions mass was slowed down, as P-RGO was about to reach its saturation adsorption for lead ions. Therefore, the saturated adsorption capacity of P-RGO for lead ions could be more than 184.5 mg g−1. The adsorption capacity of lead ions on various adsorbents was listed on Table S2.† Among these adsorbents, the adsorption capacity of P-RGO was the highest. | ||
| Fig. 6 Effects of contact time on the adsorption capacities of lead ions onto reduced graphene oxide (RGO) and pellet-like reduced graphene oxide (P-RGO). | ||
 | ||
| Fig. 7 (a) Effect of lead ion initial concentration on the adsorption capacity of reduced graphene oxide (RGO) and pellet-like reduced graphene oxide (P-RGO) (m/V = 0.2 g L−1, pH = 5.6, T = 25 °C). (b) Effect of pH value on the adsorption ratio of RGO and P-RGO (m/V = 0.2 g L−1, CLead ions = 50 mg L−1, T = 25 °C). | ||
The pH dependence of the adsorption behavior was shown in Fig. 7b. For RGO, the adsorption ratio increased and then declined with pH increasing from 1 to 6, and come to a plateau at pH = 4. However, the adsorption ratio of P-RGO increased as decreasing pH from 4 to 1, but increased and then decreased with pH from 4 to 5.6 and from 5.6 to 6, respectively. Thought the adsorption ratio of HGS on pH 1.0 was more that on pH 5.6, the consumption of acid was also increased, so the optimal pH 5.6 was selected for lead removing. The lead ions in alkaline solution would be precipitated as Pb(OH)2, so the alkaline range was not shown. Three kinds of mechanisms were proposed to facilitate the understanding of lead ions adsorption, including electrostatic interactions, ions-exchange, and complex formations.41 But the reduction degree of P-RGO was higher than that of RGO (see Fig. S3,† the XRD spectra of RGO and P-RGO), the content of oxygen-containing groups on P-RGO was less than that on RGO, so the ions-exchange was not the main mechanism for the adsorption of lead ions on P-RGO. Because the trend of Zeta potential vs. pH curves (Fig. S4†) was opposite to that of adsorption efficient vs. pH curves (Fig. 7b), the electrostatic interaction between the negatively charged P-RGO and the positively charged lead ions was the main mechanism for lead ions removal.
From an economic and environmental standpoint, an ideal adsorbent should possess not only high adsorption capacity, but also better desorption performance for the regeneration of the adsorbents. The desorption efficiency was investigated by immersing P-RGO-Pb2+ into ultrapure water (Fig. 8a). It was interesting to found that almost 80% of lead ions could be desorbed from P-RGO into water (pH = 7.0) within 10 min, i.e., the adsorbent was regenerated, while scarcely any lead ions desorbed for RGO at the same conditions. Five consecutive cycles of adsorption–desorption experiments were also carried out by using 50 mg L−1 of lead ions, as shown in Fig. 8b. The adsorption capacity decreased obviously in initial three cycles, after that, the adsorption capacity was still more than 81.6 mg g−1 and tended to be stable. Such adsorption capacity was enough for P-RGO as an adsorbent to remove lead ions from water.
 | ||
| Fig. 8 Desorption efficient of pellet-like reduced graphene oxide toward lead ions at different time in aqueous solution (a). Adsorption capacity of lead ions on the P-RGO during five consecutive cycles of desorption–adsorption (CLead ions = 50 mg L−1, T = 25 °C) (b). | ||
When P-RGO was used in the pH range of 1–4, an other mechanism, i.e., a competitive process (Fig. 9), was proposed for lead ions adsorption and desorption. Although the structure of RGO was transformed into P-RGO, π electron delocalization of graphene layers was existed and then lead ions in aqueous solution were prone to form electron-donor acceptor complexes through electrostatic interaction.11,42 The higher H+ concentrations could cause the stronger charge repulsion, and then more lead ions could permeated through graphene into the inner cavity among layers, while water molecules were almost blocked at outer layer as reduced graphene oxide was a hydrophobic material. When at higher pH value, the species of lead ions were dominant as ion association complexes with water molecules in the desorption process. These two behaviors were attributed to a capillary-like high pressure acting on ions inside graphene capillaries, which were similar as the anomalously fast permeation of precise and ultrafast molecular sieving through graphene oxide membranes.39 The graphene materials, if immersed in water, could be acted as molecular sieves, blocking all solutes with hydrated radii larger than 0.45 nm. The ionic radius of lead ions at different coordination numbers was range from 0.119–0.149 nm (Table 1). The lead ions in aqueous solution could permeate through the graphene layers at rates thousands of times faster than what was expected for simple diffusion. This was the reason for the fast adsorption and desorption of lead ions on P-RGO. Lead ions could be adsorbed on graphene sheets via ionic exchange. However, because of different affinities for water and lead ions, lead ions could be drawn and stored into the cavity in the P-RGO for its pellet like structure, which could be said as “ions storage effect”. Such effect led to higher adsorption capacity and abnormal adsorption behavior. As can be seen from the above, lead ions adsorption and desorption behaviors were affected by the structure of graphene. Pellet like structure of reduced graphene oxide could act as ions storage and then the performance of heavy metal removal was improved.
 | ||
| Fig. 9 Schematic diagrams for illuminating the lead ions behavior on pellet-like reduced graphene oxide. | ||
| Coordination number | 6 | 8 | 10 | 12 |
| Ionic radius (nm) | 0.119 | 0.129 | 0.140 | 0.149 |
Graphene-based materials had emerged as a rapidly rising star in the field of material science. Its large surface area and corresponding large adsorption capacity allowed graphene to act as a sink for various dyes and heavy metal ions, e.g., lead.11,35 Compared with the well dispersed graphene oxide, reduced graphene oxide was more welcomed due to its segregation and no secondary pollution. The results in this study showed that the influences of RGO structures on lead ions adsorption onto the RGO with different structures. The P-RGO exhibited high adsorption capacities, high adsorption and desorption rate, and strongly pH-dependent. Additionally, the lower pH value (from 1 to 4) of the solution resulted in higher lead ions removal efficiency for P-RGO, which was opposite to layered RGO. The acidic wastewater could be directly removed by P-RGO without adjusting to the neutral pH range, thus offering potential applications toward metal ions removal for environmental remediation.
4 Conclusions
A simple, one-pot reaction, electrostatic assembly method for the synthesis of P-RGO was presented. The as prepared P-RGO exhibited large adsorption capacity, high adsorption and desorption rate, and strongly pH-dependent. The outer graphene walls, if immersed in water, could be used as molecular sieves for lead ion removal.Acknowledgements
This work was supported by the National Natural Science Foundations of China (No. 20775067, 20977074, 21175115 and 21475055), the Science & Technology Committee of Fujian Province, China (No. 2012Y0065), and the Program for New Century Excellent Talents in University (NCET-11 0904).References
- Y. H. Li, S. G. Wang, J. Q. Wei, X. F. Zhang, C. L. Xu, Z. K. Luan, D. H. Wu and B. Q. Wei, Chem. Phys. Lett., 2002, 357, 263 CrossRef CAS.
- S. X. Li, L. H. Chen, F. Y. Zheng and X. G. Huang, J. Agric. Food Chem., 2014, 62, 7050 CrossRef CAS PubMed.
- T. X. Tu, S. X. Li, L. H. Chen, F. Y. Zheng and X. G. Huang, Aquat. Toxicol., 2014, 155, 269 CrossRef CAS PubMed.
- S. X. Li, L. X. Lin, F. Y. Zheng and Q. X. Wang, J. Agric. Food Chem., 2011, 59, 822 CrossRef CAS PubMed.
- G. P. Rao, C. Lu and F. Su, Sep. Purif. Technol., 2007, 58, 224 CrossRef CAS PubMed.
- M. Ghaedi, A. Shokrollahi, K. Niknam, E. Niknam, A. Najibi and M. Skylark, J. Hazard. Mater., 2009, 168, 1022 CrossRef CAS PubMed.
- O. D. Uluozlu, M. Tuzen, D. Mendil and M. Soylak, J. Hazard. Mater., 2010, 176, 1032 CrossRef CAS PubMed.
- M. Soylak, Y. E. Unsal, N. Kizil and A. Aydin, Food Chem. Toxicol., 2010, 48, 517 CrossRef CAS PubMed.
- M. M. Rao, D. K. Ramana, K. Seshaiah, M. C. Wang and S. W. C. Chien, J. Hazard. Mater., 2009, 166, 1006 CrossRef CAS PubMed.
- V. K. Gupta and I. Ali, J. Colloid Interface Sci., 2004, 271, 321 CrossRef CAS PubMed.
- Z. H. Huang, X. Zheng, W. Lv, M. Wang, Q. H. Yang and F. Kang, Langmuir, 2011, 27, 7558 CrossRef CAS PubMed.
- Q. Peng, J. Guo, Q. Zhang, J. Xiang, B. Liu, A. Zhou, R. Liu and Y. Tian, J. Am. Chem. Soc., 2014, 136, 4113 CrossRef CAS PubMed.
- A. K. Geim and K. S. Novoselov, Nat. Mater., 2007, 6, 183 CrossRef CAS PubMed.
- A. A. Balandin, S. Ghosh, W. Z. Bao, I. Calizo, D. Teweldebrhan, F. Miao and C. N. Lau, Nano Lett., 2008, 8, 902 CrossRef CAS PubMed.
- G. Eda and M. Chhowall, Adv. Mater., 2010, 22, 2392 CrossRef CAS PubMed.
- C. Lee, X. D. Wei, J. W. Kysar and J. Hone, Science, 2008, 321, 385 CrossRef CAS PubMed.
- Y. W. Zhu, S. Murali, W. W. Cai, X. S. Li, J. W. Su, J. R. Potts and R. S. Ruo, Adv. Mater., 2010, 22, 3906 CrossRef CAS PubMed.
- M. Q. Zhao, Q. Zhang, J. Q. Huang, G. L. Tian, J. Q. Nie, H. J. Peng and F. Wei, Nat. Commun., 2014, 5, 3410 Search PubMed.
- D. Li, M. B. Muller, S. Gilje, R. B. Kaner and G. G. Wallace, Nat. Nanotechnol., 2008, 3, 1170 Search PubMed.
- X. W. Yang, L. Qiu, C. Cheng, Y. Z. Wu, Z. F. Ma and D. Li, Angew. Chem., Int. Ed., 2011, 50, 7325 CrossRef CAS PubMed.
- Z. P. Chen, W. C. Ren, L. B. Gao, B. L. Liu, S. F. Pei and H. M. Cheng, Nat. Mater., 2011, 10, 424 CrossRef CAS PubMed.
- X. H. Cao, Y. M. Shi, W. H. Shi, G. Lu, X. Huang, Q. Y. Yan, Q. C. Zhang and H. Zhang, Small, 2011, 7, 3163 CrossRef CAS PubMed.
- L. G. De Arco, Y. Zhang, C. W. Schlenker, K. Ryu, M. E. Thompson and C. W. Zhou, ACS Nano, 2010, 4, 2865 CrossRef PubMed.
- Z. Xu and C. Gao, Nat. Commun., 2011, 2, 571 CrossRef PubMed.
- F. Gunes, H. J. Shin, C. Biswas, G. H. Han, E. S. Kim, S. J. Chae, J. Y. Choi and Y. H. Lee, ACS Nano, 2010, 4, 4595 CrossRef CAS PubMed.
- J. F. Shen, Y. Z. Hu, C. Li, C. Qin, M. Shi and M. X. Ye, Langmuir, 2009, 25, 6122 CrossRef CAS PubMed.
- J. L. Vickery, A. J. Patil and S. Mann, Adv. Mater., 2009, 21, 2180 CrossRef CAS PubMed.
- X. T. Zhang, Z. Y. Sui, B. Xu, S. F. Yue, Y. J. Luo, W. C. Zhan and B. Liu, J. Mater. Chem., 2011, 21, 6494 RSC.
- M. A. Worsley, P. J. Pauzauskie, T. Y. Olson, J. Biener, J. H. Satcher Jr and T. F. Baumann, J. Am. Chem. Soc., 2010, 132, 14067 CrossRef CAS PubMed.
- H. Y. Sun, Z. Xu and C. Gao, Adv. Mater., 2013, 25, 2554 CrossRef CAS PubMed.
- S. Yang, X. Feng, S. Ivanovici and K. Müllen, Angew. Chem., Int. Ed., 2010, 49, 8408 CrossRef CAS PubMed.
- K. Sohn, Y. J. Na, H. Chang, K. M. Roh, H. D. Jang and J. Huang, Chem. Commun., 2012, 48, 5968 RSC.
- S. Li, J. Zheng, D. Chen, Y. Wu, W. Zhang, F. Zheng, J. Cao, H. Ma and Y. Liu, Nanoscale, 2013, 5, 11718 RSC.
- J. Hong, K. Char and B. S. Kim, J. Phys. Chem. Lett., 2010, 1, 3442 CrossRef CAS.
- G. K. Ramesha, A. Vijaya Kumara, H. B. Muralidhara and S. Sampath, J. Colloid Interface Sci., 2011, 361, 270 CrossRef CAS PubMed.
- P. Bradder, S. K. Ling, S. B. Wang and S. M. Liu, J. Chem. Eng. Data, 2011, 56, 138–141 CrossRef CAS.
- S. B. Wang, H. Q. Sun, H. M. Ang and M. O. Tadé, Chem. Eng. J., 2013, 226, 336–347 CrossRef CAS PubMed.
- Y. J. Yao, S. D. Miao, S. Z. Liu, L. P. Ma, H. Q. Sun and S. B. Wang, Chem. Eng. J., 2012, 184, 326–332 CrossRef CAS PubMed.
- R. K. Joshi, P. Carbone, F. C. Wang, V. G. Kravets, Y. Su, I. V. Grigorieva, H. A. Wu, A. K. Geim and R. R. Nair, Science, 2014, 343, 752 CrossRef CAS PubMed.
- W. S. Hummers and R. E. Offeman, J. Am. Chem. Soc., 1958, 80, 1339 CrossRef CAS.
- L. Hao, H. Song, L. Zhang, X. Wan, Y. Tang and Y. Lv, J. Colloid Interface Sci., 2012, 369, 381 CrossRef CAS PubMed.
- M. Machida, T. Mochimaru and H. Tatsumoto, Carbon, 2006, 44, 2681 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c5ra06493g |
| ‡ Present address: Department of Chemistry & Environmental Science, Minnan Normal University, Zhangzhou, 363000, China. |
| This journal is © The Royal Society of Chemistry 2015 |