
Template size matched film thickness for effectively in situ surface imprinting: a model study of glycoprotein imprints
Gang Wu†
ab,
Jinyang Li†ab,
Xue Qu*ab,
Yuxin Zhangab,
Hua Hongab and
Changsheng Liu*ab
aThe State Key Laboratory of Bioreactor Engineering, East China University of Science and Technology, Shanghai 200237, PR China. E-mail: quxue@ecust.edu.cn; liucs@ecust.edu.cn
bKey Laboratory for Ultrafine Materials of Ministry of Education, East China University of Science and Technology, Shanghai 200237, PR China
First published on 20th May 2015
Abstract
Precisely controlling the material structure is a high requirement for biological target imprinting. In situ surface imprinting immobilized templates can offer thin-film molecular imprints containing site-directed binding sites on substrates, and therefore is appropriate for biological targets. However, the correlation between the required film thickness for superior imprinting effect and the bulk structure of biological template is not clearly understood. Here we use a series of glycoprotein imprinted films as a model to give a semi-quantitative description for their correlation. Glycoproteins with distinguished molecular sizes including ribonuclease B, glucose oxidase and horseradish peroxidase were used as templates. Covalently immobilizing glycoproteins was achieved by using m-aminophenylboronic acid modified SiO2 or Fe3O4 surface. Dopamine was polymerized onto this surface for glycoprotein imprinting. Varying polymerization time provided a series of thickness tunable imprinting films in nanometer-scale. The binding isotherm study for each glycoprotein imprints with different film thickness was performed. The optimal film thickness for the highest binding capacities and imprinting factors shows a positive correlation with its template size. The each optimized glycoprotein imprints can recognize their template in a simple or complex environment. These results suggest that the thickness of imprinted film should be tailored for matching the geometric size of fixed templates, and reveal the substantial influence of template structure on imprints design.
Introduction
Recent advances in materials science have improved the development of intelligent materials with various functions and performances. Molecular imprinting is a generic method to produce such materials with “molecular memory” by performing a polymerization of suitable functional monomer in the presence of a template molecule. Accordingly, molecularly imprinted polymers (MIP) have been utilized in many applications that require molecular recognition.1–6 This procedure takes a snapshot to the preassembly of the functional monomer and template. After the template removal, the resultant “imprinting cavities” recognize the template molecule specifically, since ideally each of the recognition cavities would have a specific shape that match the geometry of the template quite well, as well as have oriented functional groups complementary to those of the template. The “Key-lock” theory proposed by Emily Fisher can help understand this size matching relationship vividly.Although creating imprints towards small molecules is straightforward now, imprinting of larger, more delicate biological targets such as protein, DNA and even whole cells is still a challenge.7–11 The highly cross-linked polymers seriously hinder the mass transfer of biological templates and lead to the poor rebinding efficiency. The structural and conformational sensitivity of these targets to the harsh imprinting conditions is the further reason why simple extrapolation of the well established imprinting technologies for small molecules does not work.
In situ imprinting a template immobilized surface allows creating thin-film like polymer structures containing site-directed imprinting cavities which are situated at or close to the surface of imprinted polymers, thus can improve the mass transfer, as well as offer clear cut imprinting cavities.12–17 Typically, this approach involves templates anchoring, polymer film deposition in the inter-template voids and subsequent templates removal steps. In order to achieve a highly imprinting efficiency, recent works in this field have focused on the development of novel template immobilization methods,18 the design of nano-scaled supports with versatile features19–21 and the combination of different functional monomers for conferring multiple advantages.22
However, if we take a close look at successful imprints using this technique, we may discover controlling growth of the imprinted films over supports also plays an important role for generation of effective imprinting cavities. It is an easily acceptable belief that the thickness of imprinted film should be compatible with the geometric size of fixed templates. A too thin imprinted film is prone to result in shallow cavities that cannot provide enough contact area for remembering the shape of the templates precisely. An opposite case is a too thick film, for which the extraction of templates from the polymer for yielding empty imprinting cavities is hardly achieved, since almost all the template molecules are buried by polymer matrix permanently. However, to our best knowledge previous works related to surface imprinting for biomacromolecules seldom paid attention to this point, except for Zhang's23 and Liu's24,25 reports where the desired imprinting film thicknesses for targets rebinding were tailored.
In this paper, we performed a semi-quantitative study to understand the correlation between required thickness of deposited imprinted film and the geometric size of anchored biological template. Glycoproteins were used as model templates,26–29 since they have great biological and clinical importance.
The proposed surface imprinting model is illustrated in Scheme 1. Nanoparticles i.e. SiO2 and Fe3O4 were employed as solid substrates, since they are functional colloidal particles with a number of notable advancements.30–33 Glycoproteins i.e. ribonuclease B (RNase B, Mw = 14.7 kDa, 3.1 × 5.1 × 7.6 nm),34 glucose oxidase (GOx, Mw = 80 kDa, 5.8 × 13.2 × 15.1 nm)35 and horseradish peroxidase (HRP, Mw = 40 kDa, 4.0 × 6.7 × 11.7 nm)36 were used as model templates. They offer distinguished molecular sizes for easy comparation of optimal film thickness for size-different targets. The imprinting process included three steps. First, covalently immobilizing glycoprotein templates was achieved by exposing these molecules to m-aminophenylboronic acid (APBA) modified substrate surface.37 Boronic acid is reported to covalently bind cis-diol-containing molecules such as sugars and glycoproteins in basic pHs, while release them in acidic pHs. This reversible binding characteristic is employed by several groups for glycoprotein imprinting.26–29,38–41 Following that, an imprinted film of polydopamine (PDA) was deposited onto the substrate surface by inducing a self-polymerization of dopamine from its basic solution. The choice of dopamine as the functional monomer was motivated by (i) its mild aqueous polymerization environment well compatible with proteins; (ii) the available –OH and –NH2 groups offering a high number of favorable weak interactions with amino acids on the protein;32 (iii) as the most important reason, the readily controlled polymerization process by varying the reaction time, thus allows the yielding of polymer films with tunable thickness. Finally, after the PDA film formation, simple washing with acidic buffer containing 0.1% sodium dodecyl sulfate (SDS) removed the protein template and generated imprinting cavities. We estimated the binding ability as the function of film thickness as well as target selectivity of each glycoprotein imprints.
 | ||
| Scheme 1 In situ surface imprinting of immobilized glycoproteins in a film thickness controlled manner. The thickness of imprinted film needs to be tailored for matching individual glycoprotein templates. | ||
We show the proof-of-concept that indeed the thickness of imprinted film generated over substrates needs to be tailored for individual templates. The optimal film thickness for the highest binding capacities and imprinting factors shows a positive correlation with its template size.
Experimental
Materials
Dopamine, aminophenylboronic acids (APBA), hexanedioyl chloride were purchased from Adamas Chemical Company. KH550, RNase B (Mw = 14.7 kDa, pI = 8.9) were purchased from Aladdin Industrial Corporation. HRP (pI = 3.9–9.0, Mw = 40 kDa), GOx (pI = 4.6, Mw = 80 kDa), bovine serum albumin (BSA) (pI = 4.9, Mw = 66 kDa) and haemoglobin (Hb) (pI = 6.9, Mw = 64 kDa) were purchased from Yuanye Biotech Ltd. (Shanghai, China). FeCl3·6H2O, NaAc, ethylene glycol, diethylene glycol were all purchased from Jingchun Reagent Co., Ltd. (Shanghai, China).SiO2 nanoparticles (ca. 224 nm in diameter; 2.2 g cm−3 in density) were from Dongjian Biotech Ltd. (Nanjing, China). Fe3O4 nanoparticles (ca. 253 nm in diameter; 5.18 g cm−3 in density) were synthesized according to a modified hydrothermal reaction.42,43 All other reagents were purchased from China Nation Medicines (Shanghai, China).
Synthesis of APBA modified nanoparticles
The synthesis of APBA modified Fe3O4 involved two steps including preparation of amine-functionalized magnetic particles and surface modification. (1) For amine modified Fe3O4, 3 g of Fe3O4 was first dispersed into ethanol–H2O mixture solution (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) adjusted at pH 4 by acetic acid, and then added with 4 mL of KH550. The mixture solution was stirred for 12 h at 60 °C with Ar protection for amino group grafting, followed by a sufficient washing, vacuum drying and weighing. (2) For APBA surface modification, 100 mg amine-functionalized Fe3O4 was dispersed into 30 mL of tetrahydrofuran under continuous mechanically stirring, 0.2 mL of hexanedioyl chloride was added into the mixture by injection. After reaction for 4 h, the product was collected with the help of a magnet and washed sufficiently to remove the excess hexanedioyl chloride, and then redispersed into 30 mL of dry chloroform. Into the resultant mixture, 0.2 g of 3-aminobenzeneboronic acid was added. After reaction for 6 h under stirring, the product was collected and washed according to the above mentioned procedure to remove excess 3-aminobenzeneboronic acid, and then vacuum dried to obtain the APBA-functionalized Fe3O4 nanoparticles. The APBA modified SiO2 were prepared in the same manner.
:1) adjusted at pH 4 by acetic acid, and then added with 4 mL of KH550. The mixture solution was stirred for 12 h at 60 °C with Ar protection for amino group grafting, followed by a sufficient washing, vacuum drying and weighing. (2) For APBA surface modification, 100 mg amine-functionalized Fe3O4 was dispersed into 30 mL of tetrahydrofuran under continuous mechanically stirring, 0.2 mL of hexanedioyl chloride was added into the mixture by injection. After reaction for 4 h, the product was collected with the help of a magnet and washed sufficiently to remove the excess hexanedioyl chloride, and then redispersed into 30 mL of dry chloroform. Into the resultant mixture, 0.2 g of 3-aminobenzeneboronic acid was added. After reaction for 6 h under stirring, the product was collected and washed according to the above mentioned procedure to remove excess 3-aminobenzeneboronic acid, and then vacuum dried to obtain the APBA-functionalized Fe3O4 nanoparticles. The APBA modified SiO2 were prepared in the same manner.
Preparation of the Fe3O4@MIP-t h and Fe3O4@NIP-t h
80 mg of APBA modified Fe3O4 nanoparticles and 20 mg of HRP were first added into 40 mL of Na2CO3–NaHCO3 buffer (10 mM, pH 9.6). The mixture solution was mechanical stirred for 2 h at room temperature for HRP binding. And then they were collected and exposed to a dopamine solution (1 mg mL−1). The polymerization of dopamine was allowed for t h (1, 3, 6, 9, 15) at room temperature. After the reaction, the product was collected with a magnet and washed with a 3% (v/v) solution of acetic acid containing 0.1% (w/v) SDS for five times, then thoroughly washed with distilled water until no HRP could be detected from the washing solvent by monitoring the absorbance at 405 nm with UV-vis spectrometer (Bradford assay was used for monitoring RNase B and GOx washing). The Fe3O4@RNase B-MIP-t h and Fe3O4@GOx-MIP-t h were prepared and washed using the same recipe but with template RNase B and GOx. The control of Fe3O4@NIP-t h were prepared and washed using the same recipe but without the addition of the protein template.Preparation of the SiO2@HRP-MIP-t h and SiO2@NIP-t h
The SiO2@HRP-MIP-t h and SiO2@NIP-t h were prepared with the same procedure as that for Fe3O4@MIP-t h and Fe3O4@NIP-t h, except that the added APBA modified SiO2 nanoparticles was controlled at 31 mg for offering a comparable surface area to that of Fe3O4, which is based on a simple geometric calculation.Binding experiments
Briefly, 10 mg of glycoprotein imprinted particles or control of non-imprinted particles were suspended in 1.0 mL of protein template standard solutions (in pH 9.6 Na2CO3–NaHCO3 buffer) with different initial concentrations, respectively. The samples were incubated on a rocking table for 12 h at room temperature and then separated with a magnet. The concentration of free template protein in the supernatant was measured by UV-vis spectroscopy at 405 nm for HRP and by Bradford assay for RNase B and GOx (Bradford Protein Assay Kit), respectively.The selectivity of the imprinted particles was investigated using BSA, Hb and the other two non-templated glycoproteins as reference.
The binding capacity (Q) of the template protein or competitive protein bound to the imprinted polymers is defined as:
 | (1) |
The imprinting factor (IF) is calculated from the following equation:
 | (2) |
APBA–HRP interaction analysis by ITC
ITC was employed to study the interactions between HRP and boronic acid in different pHs. The experiments were performed using microcalorimeter Microcal ITC 200. Na2CO3–NaHCO3 buffer (10 mM, pH = 9.6), HAc–NaAc buffer (pH = 5.0) were filtered, thoroughly degassed, and used for preparing the HRP and aminophenylboronic acid solutions. In each titration, 1.5 μL of aminophenylboronic acid solution was injected into the HRP solution with a time interval of 2 min at 25 °C. The heat evolved during the binding process was measured. The data were fitted to a one-site binding model using a nonlinear least-squares procedure and give the binding affinity Ka and enthalpy ΔH.Characterization
The morphology and structure of Fe3O4 and SiO2 nanoparticles were examined by transmission electron microscope (JEM-2100F, JEOL, Japan), scanning electron microscopy (S-4800, Hitachi, Japan) and Atomic Force Microscope (Veeco/DI, USA). The infrared spectra were recorded on Fourier transform infrared (FT-IR) spectrometer (Nicolet 6700, USA). Electrophoretic analysis of proteins was performed using regular SDS-PAGE (Bio-Rad, Hercules, CA) with 12% running and 5% stacking gels. Proteins were stained with Coomassie Brilliant Blue G250.Results and discussion
pH-dependent glycoprotein immobilization by boronic acid
We performed isothermal titration calorimetry (ITC) to demonstrate the pH dependent covalently binding between boronic acid and glycoprotein of HRP. The first titration experiment was done by adding APBA to a solution of HRP at pH 9.6. Fig. 1A shows the typical heat release for boronic acid-HRP binding. The calculated thermodynamic properties demonstrate a relatively high binding constant (4.9 × 103 M−1) and favourable enthalpy (−2.0 kcal mol−1). The second titration was performed by adding APBA to a solution of HRP at pH 5.0. The integrated heat evolution curve in Fig. 1B became featureless, suggesting no binding occurred in this acidic condition. The mechanism of HRP–boronic acid interactions can be interpreted by the equilibrium formation of a heterocyclic diester from 1,2-diol groups on HRP sugar chains and a tetrahedral boronate ion, which is a function of the ionization constant of Ka of the boronic acid moiety (Fig. 1C).44 The boronate ion is responsible for the active binding and decreased [H+] facilitates the generation of boronate ions. The pKa value for APBA is reported at 8.7–8.9,45,46 therefore favorable HRP–APBA interactions would occur when media pH is over that value. Our observation that the boronic acid–HRP binding is preferable in the case of pH 9.6 over an acidic pH of 5.0 shows good agreement with this principle. | ||
| Fig. 1 pH-dependent glycoprotein binding by boronic acid group on APBA. (A and B) Isothermal titration calorimetry (ITC) measurement shows exotherms for the titration of APBA into HRP solution at pH 9.6, while titration at pH 5.0 shows small exotherms. (C) Depiction of APBA–sugar binding equilibrium. The binding for sugar is facilitated at a basic pH that results in conversion of boronic acid species into the boronate ions. (D) Photographs show that mixing TMB solution for 2 h with APBA modified Fe3O4 particles binding HRP at pH 9.6 yields blue color (left), while that with particles binding HRP at pH 5.0 does not cause color changes (right) (presumably APBA modified Fe3O4 particles bind more HRP at basic pH than those at acidic pH). | ||
We further provide visual evidence for pH dependent immobilization of HRP on boronic acid modified substrate. Fe3O4 was first modified with APBA and then exposed to HRP solutions at pH 9.6 and pH 5.0 respectively to allow HRP binding. After that, the particles were collected and washed substantially by water. The presence of HRP on Fe3O4 was determined in situ by enzyme activity assay. Briefly, the particles were dispersed into tetramethylbenzidine dihydrochloride (TMB) staining solutions to initiate colorimetric reaction. After 2 h incubation, the blue color was observed in TMB solution that was contacted with APBA–Fe3O4 particles binding HRP at pH 9.6 (Fig. 1D left), while the contact with particles binding HRP at pH 5.0 did not cause any solution color changes (Fig. 1D right). This may indicate APBA–Fe3O4 particles bind more HRP at basic pH than those at acidic pH: an obvious pH-dependent binding performance similar to the free APBA molecules.
These results demonstrate the boronic acid group can covalently immobilize glycoprotein templates at basic pH (the imprinting condition) and release them at acid pH (the template washing condition), which is consistent with other groups' findings.22,26,29
In situ surface imprinting glycoproteins by dopamine
We next initiated the surface imprinting of immobilized glycoprotein through dopamine self-polymerizing. APBA modified SiO2 particles (ca. 224 nm in diameter; ca. 2.2 g cm−3 in density) were anchored with HRP proteins at pH 9.6 and then exposed to a 1 mg mL−1 dopamine solution in air at pH 9.6 for an allotted time. Following the PDA film formation and protein templates removing, core–shell MIP particles were obtained [denoted as SiO2@HRP-MIP-t h, there t represents the imprinting time]. The collected samples show typical dark brown color from PDA. Their corresponding non-imprinted particles NIP were synthesized following the same procedure except for the addition of templates [denoted as SiO2@NIP-t h]. This process was also applied to Fe3O4 magnetic particles with a diameter of ca. 253 nm and a density of 5.18 g cm−3. We should note that the quantity of added Fe3O4 is carefully controlled to offer a comparable surface area to that of SiO2, which is based on a simple geometric calculation.Fig. 2A shows scanning electron microscopy (SEM) images of bare nanoparticles and HRP imprinted nanoparticles. After 3 h polymerization, the deposited polydopamine film was observed to mask the original appearance of the SiO2 substrates and yield a new granular surface, as shown in the upper two images in Fig. 2A. An opposite phenomenon was observed for Fe3O4 particles. The lower two images revealed that the formed PDA film seems to fill in the valleys on Fe3O4 and lead to a decrease in surface roughness.
 | ||
| Fig. 2 Characterization of polydopamine (PDA) film. (A) SEM images of nanoparticles with PDA imprinting film which is polymerized on HRP immobilized Fe3O4 or SiO2 surface for 3 h. (B) AFM images of SiO2, SiO2@HRP-MIP-3 h and SiO2@NIP-3 h (note that red square indicated area has different surface roughness for each sample, SiO2@HRP-MIP-3 h has a little rougher surface compared to SiO2@NIP-3 h presumably because of the presence of imprinting cavities created by HRP protein). (C) FTIR spectra for APBA, Fe3O4 (APBA modified) and Fe3O4@NIP-3 h (dopamine is polymerized on Fe3O4 surface for 3 h) provide evidence for PDA deposition. | ||
We further performed atomic force microscopy (AFM) measurement to compare the morphological difference between MIP and the corresponded NIP nanoparticles. Bare SiO2 substrate is used as control. Fig. 2B shows that compared to SiO2 a rough surface for SiO2@NIP-3 h was yielded after the deposition of PDA. While for HRP imprinted sample of SiO2@HRP-MIP-3 h, the surface roughness seems more remarkable. Quantitative analysis of local surface topography (red square indicated area) for each sample was achieved by using Nanoscope image processing software. The roughness parameters (roughness average Ra and roughness root mean square RMS) listed below provide quantitative evidence that the HRP imprints contain a little rougher surface, which is presumably because of the presence of imprinting cavities created by HRP protein.
Chemical evidence for the formation of polydopamine film was obtained by comparing Fourier-transform infrared spectroscopy (FTIR) spectra for APBA modified nanoparticles of APBA–Fe3O4 and polydopamine further deposited sample of Fe3O4@NIP-3 h. Fig. 2C shows that the spectrum for Fe3O4@NIP-3 h shows a new peak at 1220 cm−1, which is assigned to the C–O stretching vibration of phenolic hydroxyl. New peaks around 2830–2950 cm−1 are attributed to the C–H stretching model of alkyl groups. These results provide chemical evidence for polydopamine introducing onto the nanoparticles.
The controlled growth of PDA film
Several publications have reported that the thickness of surface deposited PDA film can be controlled by the concentration of dopamine, temperature, solution pH, polymerization time and number of polymerization cycles.47,48 Herein, we grow thickness controlled PDA film by varying polymerization time.We first estimated the PDA growth as a function of polymerization time on protein imprinted SiO2 nanoparticles. The polymerization of dopamine on HRP template fixed surface was allowed to proceed for a predetermined time interval, and the yielded core–shell particles were imaged by transmission electron microscope (TEM). The results in Fig. 3A show that the deposited PDA forms a well-defined shell on the outside of SiO2. The evolution of PDA thickness with time elongation was quantified. At least 10 different particles for one sample were measured to obtain an average of film thickness. The results are summarized in Fig. 3D. The PDA film was observed to grow rapidly in the beginning, followed by a slow second step.
 | ||
| Fig. 3 The film-thickness controlled PDA growth on substrate along with polymerization time increasing. (A–C) TEM images of PDA on HRP immobilized SiO2, HRP immobilized Fe3O4 and bare SiO2 (APBA modified). (D) Plot of the PDA film thickness versus polymerization times. (note the fed nanoparticles provide comparable surface areas) [data represent mean values ± SD (n = 10)]. These results indicate that the PDA growth is not influenced by the presence of protein template or by the contacted substrate chemistry. Controlling PDA film thickness on solid substrates can be achieved by varying polymerization time. | ||
We next performed a parallel study on HRP fixed Fe3O4 surface to investigate the substrate influence on PDA formation. The quantity of fed Fe3O4 was controlled to offer a comparable surface area to SiO2. TEM observations in Fig. 3B demonstrate the formation of PDA, and quantitative description of film growth in Fig. 3D displays a similar PDA growth tendency with that on SiO2. These results suggest that the growth of PDA would be not influenced by the substrate chemistry.
We further estimated the protein template influence on PDA formation by comparing PDA film thickness on HRP fixed SiO2 and bare SiO2 (APBA modified). Briefly, the SiO2 substrates without template immobilization were exposed to the dopamine solution for allotted time and the film thickness was determined by TEM imaging. The similar film growing behavior to that on HRP presented surface was observed, as shown in Fig. 3C and D, which suggests that the presence of globular templates would not take up too much space in the imprinting plane.
In summary, these results demonstrate that the PDA growth is not influenced by the presented protein template or by the contacted substrate chemistry. Controlling PDA film thickness on solid substrates can be easily achieved by a simply adjustment on reaction time.
Film thickness dependent imprinting effect for glycoprotein template
We performed the equilibrium binding study for MIP particles with various film thicknesses towards their individual templates. Briefly, the imprinted particles or control of non-imprinted particles prepared with different polymerization time were incubated respectively with protein template solutions with initial concentrations ranging from 0.1 to 0.8 mg mL−1 for 12 h to obtain a saturated adsorption. The particles were then separated and the adsorbed proteins were quantified by UV-vis spectroscopy for HRP or by Bradford assay for RNase B and GOx, respectively.Fig. 4 shows the binding isotherms for HRP imprints and their corresponded control of non imprinted samples. Langmuir–Freundlich (LF) isotherm was employed to model the binding curves of Fe3O4@HRP-MIP-t h (t = 1, 3, 6, 9 and 15 h) in Fig. 4A and Fe3O4@NIP-t h (t = 1, 3, 6, 9 and 15 h) in Fig. 4B (dot line in Fig. 4). LF isotherm has the desirable attribute of approximating both homogenous and heterogeneous MIP, and thus enables direct comparison of the binding properties of MIP with very different distributions of binding sites.49 The LF modeling is performed following the equation.
 | (3) |
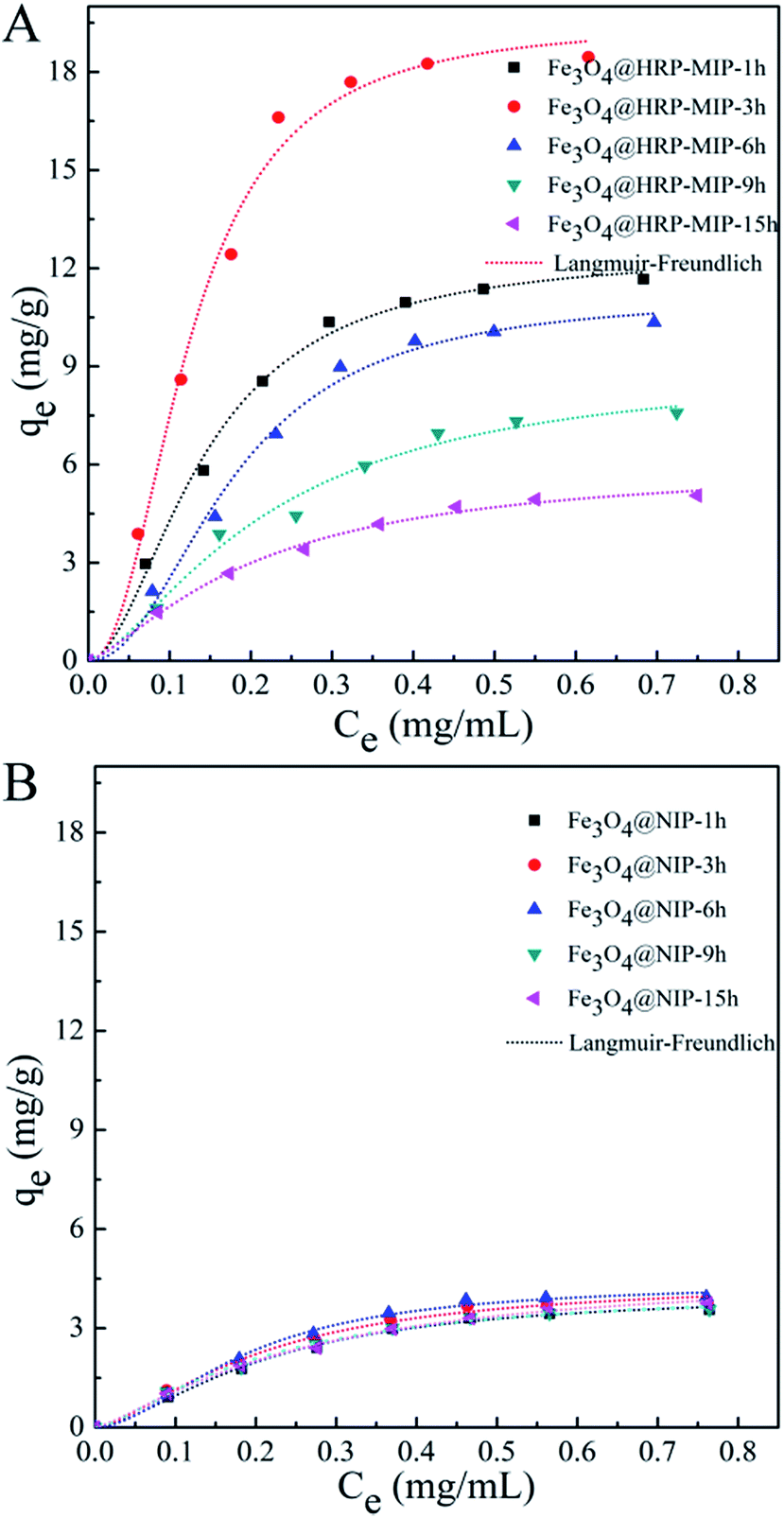 | ||
| Fig. 4 Binding isotherm plots of Fe3O4@HRP-MIP-t h (A) and Fe3O4@NIP-t h (t = 1, 3, 6, 9, 16 h) (B). The experimental data were fit to a Langmuir–Freundlich (LF) isotherm. | ||
| Sample | Fe3O4@HRP-MIP-t h | Fe3O4@NIP-t h | IF | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Q0 (mg g−1) | KLF (mL mg−1) | n | R2 | Q0 (mg g−1) | KLF (mL mg−1) | n | R2 | ||
| 1 h | 12.52 | 38.46 | 1.87 | 0.995 | 4.12 | 11.92 | 1.58 | 0.996 | 3.04 |
| 3 h | 19.50 | 110.03 | 2.24 | 0.998 | 4.49 | 11.24 | 1.52 | 0.988 | 4.34 |
| 6 h | 11.19 | 40.28 | 2.13 | 0.991 | 4.39 | 21.86 | 1.82 | 0.993 | 2.55 |
| 9 h | 9.09 | 9.64 | 1.51 | 0.982 | 4.21 | 9.81 | 1.43 | 0.995 | 2.16 |
| 15 h | 6.10 | 8.69 | 1.36 | 0.994 | 4.76 | 5.94 | 1.29 | 0.996 | 1.25 |
One can see that LF isotherm fit the binding behavior of the MIP and NIP materials quite well over the entire concentration window and gives high correlation coefficients (R2). The predicted KLF and Q0 values in Table 1 indicate that MIP of Fe3O4@HRP-MIP-t h have better HRP binding ability in contrast to NIP, which suggests the successful creation of imprinting cavities on MIP. Comparison of the binding parameters of series MIP with different polymerization time reveals that the polymerization time has a significant influence on the binding properties of these core–shell structures. An enhanced HRP binding was achieved along with the PDA deposition time increasing from 1 h to 3 h, while a further time increase up to 15 h reduced the binding ability. Fe3O4@HRP-MIP-3h has the highest association constant KLF of 110.03 mL mg−1, as well as the highest binding capacities of 19.5 mg g−1. The binding performances of various NIPs are all poor, showing no dependence on the polymerization time. A maximum imprinting factor IF of 4.34 was achieved in 3 h polymerization system. As described in Fig. 4, the elongation of polymerization time modulated a continuous growth of the PDA shell. The large structure of protein templates in contrast to small molecules does not favor a highly free molecular transfer within the MIP matrix. In this context, the time dependent binding behavior of HRP imprinted PDA film can be ascribed to polymerization time modulated film thickness variation: a thin PDA film of 5.7 nm prepared by a short polymerization time i.e. 1 h, yielded shallow imprinting cavities and presumably could not offer strong multiple interactions towards HRP, although an easy access of protein targets to these cavities could be anticipated; however an excessively thick film i.e. film prepared by 15 h polymerization has a thickness of 25.3 nm, would trap the HRP templates within PDA matrix tightly and thus hardly generate hollow binding cavities. A PDA film with an appropriate thickness of 10.2 nm by 3 h polymerization was proved to be an optimal, presumably because of the effective exposure of imprinting cavities towards HRP targets, as well as the enough functional groups being offered for HRP binding.
We further investigated the film thickness dependence of imprinting ability for RNase B imprinted nanoparticles. RNase B is used as a model template with small geometric size. The binding isotherm studies for RNase B imprinted PDA films and non imprinted controls were performed in the same manner as HRP relative systems, and the binding curves and parameter values are shown in Fig. 5 and Table 2, respectively. Fe3O4@RNase B-MIP-t h groups were observed to have higher binding abilities than their controls, and also display a polymerization time dependent binding behavior to RNase B, which is similar to the results observed for HRP imprinted PDA in Fig. 4. The maximum binding capacity of 16.73 mg g−1 and the highest association constant KLF of 96.32 mL mg−1 were achieved in Fe3O4@RNase B-MIP-1 h. Further deposition of PDA film induced an adverse effect on MIP binding ability. As a comparison, the binding curves of all the NIPs controls in Fig. 5B did not show significant adsorption to RNase B. The PDA film fabricated by 1 h polymerization represents a thin-walled structure about 5.7 nm. The observed maximum IF value of 4.39 for RNase B in this film suggests that the relatively thin film structure is appropriate for effectively imprinting templates with small size for example RNase B.
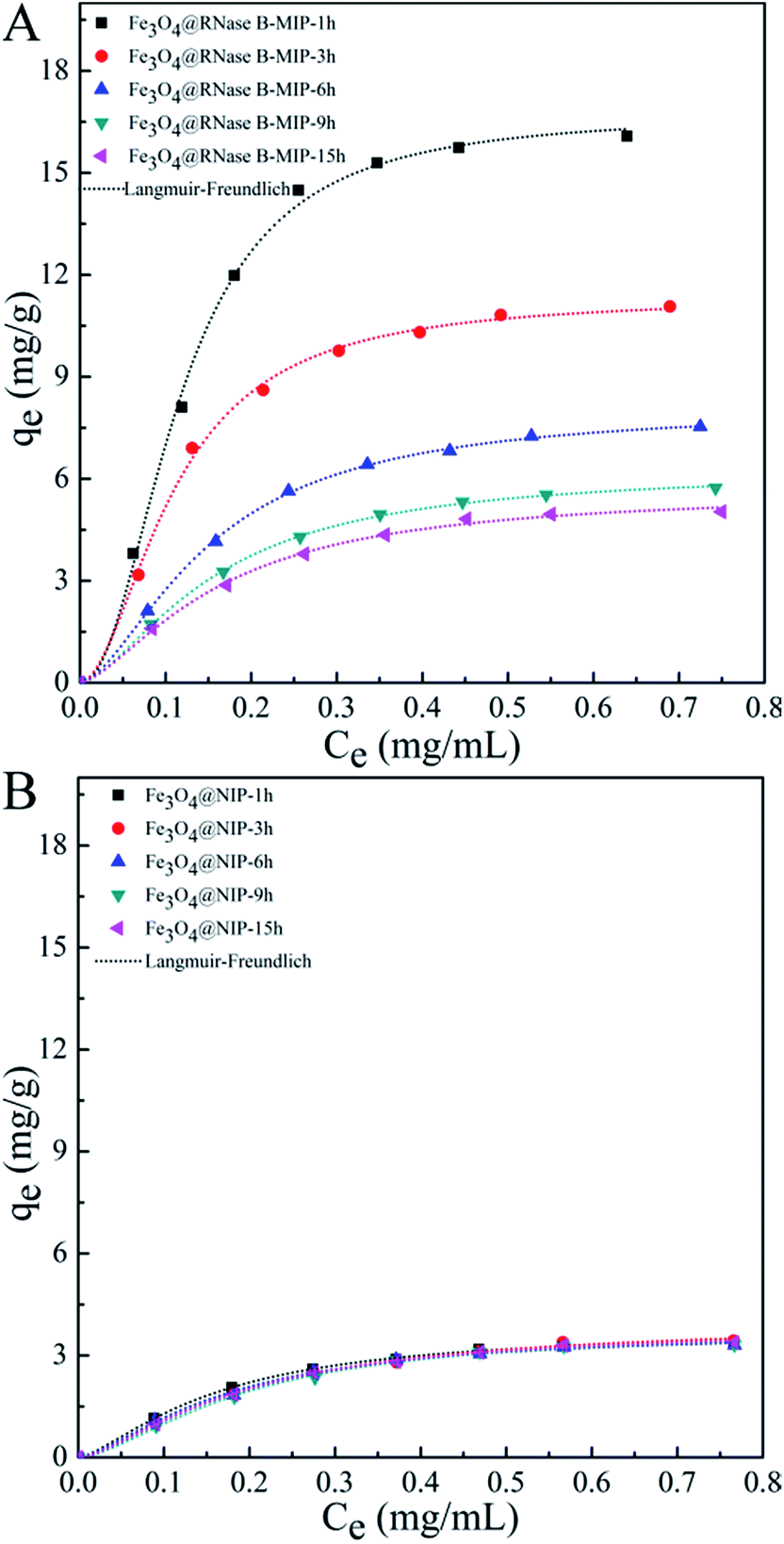 | ||
| Fig. 5 Binding isotherm plots of Fe3O4@RNase B-MIP-t h (A) and Fe3O4@NIP-t h (t = 1, 3, 6, 9, 16 h) (B). The experimental data were fit to a Langmuir–Freundlich (LF) isotherm. | ||
| Sample | Fe3O4@RNase B-MIP-t h | Fe3O4@NIP-t h | IF | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Q0 (mg g−1) | KLF (mL mg−1) | n | R2 | Q0 (mg g−1) | KLF (mL mg−1) | n | R2 | ||
| 1 h | 16.73 | 96.32 | 2.15 | 0.997 | 3.81 | 13.74 | 1.43 | 0.998 | 4.39 |
| 3 h | 11.37 | 59.73 | 1.85 | 0.997 | 3.97 | 10.97 | 1.46 | 0.997 | 2.86 |
| 6 h | 8.13 | 21.98 | 1.63 | 0.999 | 3.74 | 13.32 | 1.47 | 0.995 | 2.17 |
| 9 h | 6.27 | 18.91 | 1.58 | 0.989 | 3.78 | 13.95 | 1.59 | 0.996 | 1.66 |
| 15 h | 5.65 | 16.08 | 1.52 | 0.996 | 3.82 | 13.18 | 1.52 | 0.998 | 1.48 |
Parallel binding isotherm investigation was performed for GOx glycoprotein, a model template with large size. The results in Fig. 6 and Table 3 clearly demonstrate that the imprinting performance of the PDA film is closely related to the film thickness (i.e. polymerization time). The most desired binding capacities and IF were found in the system by 6 h polymerization (i.e. Fe3O4@GOx-MIP-6 h), where a film thickness of ∼16.3 nm can be offered, probably meet the requirement for imprinting large templates such as GOx.
 | ||
| Fig. 6 Binding isotherm plots of Fe3O4@GOx-MIP-t h (A) and Fe3O4@NIP-t h (t = 1, 3, 6, 9, 16 h) (B). The experimental data were fit to a Langmuir–Freundlich (LF) isotherm. | ||
| Sample | Fe3O4@GOx-MIP-t h | Fe3O4@NIP-t h | IF | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Q0 (mg g−1) | KLF (mL mg−1) | n | R2 | Q0 (mg g−1) | KLF (mL mg−1) | n | R2 | ||
| 1 h | 11.55 | 30.52 | 1.81 | 0.995 | 3.69 | 19.11 | 1.73 | 0.996 | 3.13 |
| 3 h | 12.30 | 52.13 | 1.96 | 0.999 | 3.38 | 25.31 | 1.85 | 0.993 | 3.64 |
| 6 h | 18.40 | 92.77 | 2.18 | 0.997 | 3.57 | 16.41 | 1.60 | 0.997 | 5.15 |
| 9 h | 9.18 | 27.86 | 1.71 | 0.998 | 3.72 | 11.56 | 1.55 | 0.991 | 2.47 |
| 15 h | 6.75 | 20.60 | 1.60 | 0.996 | 3.67 | 11.77 | 1.46 | 0.996 | 1.84 |
The film thickness dependence of KLF and IF for each variety of glycoprotein are summarized in Fig. 7. For RNase B glycoprotein with the smallest geometric size, the optimal imprinting effect was observed from PDA with a thickness of 5.7 nm, the thinnest film prepared from a 1 h polymerization (i.e. Fe3O4@RNase B-MIP-1 h). For the case of HRP glycoprotein with a medium size, a desired imprinting effect could be offered by PDA with 10.2 nm in thickness, which is a relatively thick film prepared from a 3 h polymerization (i.e. Fe3O4@HRP-MIP-3 h). Large template such as GOx however, would require a considerable thick PDA film of 16.3 nm, and therefore needs a further longer polymerization time of 6 h (i.e. Fe3O4@GOx-MIP-6 h). These results suggest that the thickness of imprinted film should match the geometric size of fixed templates quite well and reveal from one angle the substantial influence of template structure on imprints design.
 | ||
| Fig. 7 The KLF and IF evolutions as the function of PDA film thickness for each glycoprotein imprints. The film thickness of the glycoprotein imprinted PDA that has the best imprinting performance shows a positive correlation with its template size. | ||
Rebinding selectivity
We further performed the rebinding selectivity studies for optimized protein imprints. A single protein batch rebinding test was first run for HRP imprints (i.e. Fe3O4@HRP-MIP-3 h). In addition to the template of HRP, four other proteins two glycoproteins including RNase B and GOx, and two non-glycoproteins including bovine serum albumin (BSA) and hemoglobin (Hb) were used as reference. The respective proteins at a concentration of 0.5 mg mL−1 were exposed to 10 mg of Fe3O4@HRP-MIP-3 h and the control of Fe3O4@NIP-3 h. The amount of bound protein was quantified and listed in Fig. 8A. The HRP-imprinted particles clearly showed the greatest affinity for HRP, followed in order by RNase B, GOx, Hb and BSA. The obviously preferential HRP adsorption over glycoproteins i.e. RNase B and GOx, is presumably because of the presence of HRP size matched imprinting cavities, instead of solely sugar chain targeted boronate groups. The relatively less adsorbed Hb and BSA compared to glycoproteins suggest that the sugar chain–boronate interactions play an important role in the binding process. The selectivity of RNase B imprints (i.e. Fe3O4@RNase B-MIP-1 h) was determined in the same manner. The adsorption capacities of Fe3O4@RNase B-MIP-1 h and Fe3O4@NIP-1 h nanoparticles for five kinds of proteins were presented in Fig. 8B. The RNase B imprints exhibited an appreciable selectivity towards the template protein of RNase B, and favorable adsorption towards glycoproteins than non-glycoproteins. The selectivity study for GOx-imprinted particles (i.e. Fe3O4@GOx-MIP-6 h) in Fig. 8C indicates GOx imprints have template specific binding property, which is consistent with what we found for HRP and RNase B imprints systems. | ||
| Fig. 8 Rebinding selectivity studies for glycoprotein imprints in a single protein environment. (A) Adsorbed protein amount to Fe3O4@HRP-MIP-3 h and Fe3O4@NIP-3 h for different proteins. (B) Adsorbed protein amount to Fe3O4@RNase B-MIP-1 h and Fe3O4@NIP-1 h for different proteins. (C) Adsorbed protein amount to Fe3O4@GOx-MIP-6 h and Fe3O4@NIP-6 h for different proteins. The glycoprotein imprints show high selectivity towards their templates. | ||
A ternary protein competitive adsorption study was performed to provide more robust evidences for template selective binding by these glycoprotein imprints. The three glycoprotein imprints were individually contacted with a mixture solution composed of equal amount of HRP, RNase B and GOx (0.4 mg mL−1) for 12 h to obtain a saturated adsorption. After this treatment, the imprinted particles were separated and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) was used to visualize the proteins left in the solution. As shown in Fig. 9, there are three bands in the lane 1 for mixture solution, indicating GOx, HRP and RNase B, respectively. After the mixture was treated with HRP imprints, the HRP band clearly faded while the other two proteins remained almost unchanged (seeing lane 2), which indicates the specific adsorption for HRP template. The extraction of RNase B from the protein mixture by RNase B imprints is also specific, proved by the lightest band corresponding to RNase B in the lane 3. GOx imprints also display the obvious inclination for GOx binding. The lane 4 shows that the exposure of protein mixture to GOx imprints clearly reduced the GOx content in the protein mixture. In summary, both the single batch and competitive adsorption studies provide evidence for the successful fabrication of imprinting cavities for specific templates, which enable us to selectively extract the targets from a complex environment.
 | ||
| Fig. 9 Analysis of a mixture of three model glycoproteins treated with glycoprotein imprints by SDS-PAGE. Lane 1, 0.4 mg mL−1 HRP, RNase B and GOx ternary solution; lane 2, remaining solution after the treatment by Fe3O4@HRP-MIP-3 h; lane 3, remaining solution after the treatment by Fe3O4@RNase B-MIP-1 h; lane 4, remaining solution after the treatment by Fe3O4@GOx-MIP-6 h. | ||
Conclusions
We report a semi-quantitative study for understanding the correlation between the imprinting material structure and employed template size. Glycoprotein surface imprinting model was established by using boronate chemistry for template immobilizing and dopamine for imprinting. Three varieties of glycoproteins with different geometric size were imprinted, respectively. The imprinting performance including binding capacities, binding constants and imprinting factors for the as-prepared PDA imprinting films with varying thickness were estimated, and the rebinding selectivity of the optimized PDA imprinting film was also evaluated. The results indicate that similar to the “key-lock” principle running through the whole imprinting science, a template size matched imprinting film thickness is also important for achieving desired imprinting effect. Also, the PDA imprinting film with the optimal thickness shows significantly selective recognition towards its glycoprotein target, indicating an existence of effective imprinting cavities in such film structure.Acknowledgements
We acknowledge the financial support from the National Basic Research Program of China (2012CB933600), the National Natural Science Foundation of China (51103043), Innovation Program of Shanghai Municipal Education Commission (14ZZ060), Shanghai Science and Technology Development Funds (14QA1401000) and the 111 project (B14018). We also thank Dr Gregory F. Payne for data discussion.Notes and references
- C. A. Carlson, J. A. Lloyd, S. L. Dean, N. R. Walker and P. L. Edmiston, Anal. Chem., 2006, 78, 3537–3542 CrossRef CAS PubMed
.
- S. C. Zimmerman and N. G. Lemcoff, Chem. Commun., 2004, 5–14 RSC
- L. Ye and K. Mosbach, Chem. Commun., 2008, 20, 859–868 CAS
- J. F. Yin, Y. Cui, G. L. Yang and H. L. Wang, Chem. Commun., 2010, 46, 7688–7690 RSC
- S. S. Khurshid, C. E. Schmidt and N. A. Peppas, J. Biomater. Sci., Polym. Ed., 2011, 22, 343–362 CrossRef CAS PubMed
- A. Uchida, Y. Kitayama, E. Takano, T. Ooya and T. Takeuchi, RSC Adv., 2013, 3, 25306–25311 RSC
- F. L. Dickert, O. Hayden, R. Bindeus, K. J. Mann, D. Blaas and E. Waigmann, Anal. Bioanal. Chem., 2004, 378, 1929–1934 CrossRef CAS PubMed
- L. D. Bolisay, J. N. Culver and P. Kofinas, Biomaterials, 2006, 27, 4165–4168 CrossRef CAS PubMed
- M. J. Whitcombe, I. Chianella, L. Larcombe, S. A. Piletsky, J. Noble, R. Porter and A. Horgan, Chem. Soc. Rev., 2011, 40, 1547–1571 RSC
- E. P. Herrero, E. M. Valle and N. A. Peppas, Ind. Eng. Chem. Res., 2010, 49, 9811–9814 CrossRef CAS
- N. M. Bergmann and N. A. Peppas, Prog. Polym. Sci., 2008, 33, 271–288 CrossRef CAS PubMed
- T. Takeuchi and T. Hishiya, Org. Biomol. Chem., 2008, 6, 2459–2467 CAS
- T. Shiomi, M. Matsui, F. Mizukami and K. Sakaguchi, Biomaterials, 2005, 26, 5564–5571 CrossRef CAS PubMed
- J. Bognar, J. Szucs, Z. Dorko, V. Horvath and R. E. Gyucsanyi, Adv. Funct. Mater., 2013, 23, 4703–4709 CrossRef CAS PubMed
- M. Zhang, X. H. Zhang, X. W. He, L. X. Chen and Y. K. Zhang, Nanoscale, 2012, 4, 3141–3147 RSC
- D. M. Gao, Z. P. Zhang, M. H. Wu, C. G. Xie, G. J. Guan and D. P. Wang, J. Am. Chem. Soc., 2007, 129, 7859–7866 CrossRef CAS PubMed
- Y. Kamon, R. Matsuura, Y. Kitayama, T. Ooya and T. Takeuchi, Polym. Chem., 2014, 5, 4764–4771 RSC
- G. Q. Fu, H. Y. He, Z. H. Chai, H. C. Chen, J. Kong, Y. Wang and Y. Z. Jiang, Anal. Chem., 2011, 83, 1431–1436 CrossRef CAS PubMed
- Y. X. Li, M. Hong, M. Miao, Q. Bin, Z. Y. Lin, Z. W. Cai and G. N. Chen, J. Mater. Chem. B, 2013, 1, 1044–1051 RSC
- R. X. Gao, X. Kong, X. Wang, X. W. He, L. X. Chen and Y. K. Zhang, J. Mater. Chem., 2011, 21, 17863–17871 RSC
- A. V. Linares, F. Vandevelde, J. Pantigny, A. F. Cordin and K. Haupt, Adv. Funct. Mater., 2009, 19, 1299–1303 CrossRef CAS PubMed
- F. X. Gao, X. T. Ma, X. W. He, W. Y. Li and Y. K. Zhang, Colloids Surf., A, 2013, 433, 191–199 CrossRef CAS PubMed
- H. Q. Zhang, J. H. Jiang, H. T. Zhang, Y. Zhang and P. C. Sun, ACS Macro Lett., 2013, 2, 566–570 CrossRef CAS
- S. S. Wang, J. Ye, Z. J. Bie and Z. Liu, Chem. Sci., 2014, 5, 1135–1140 RSC
- X. D. Bi and Z. Liu, Anal. Chem., 2014, 86, 959–966 CrossRef CAS PubMed
- L. Li, Y. Lu, Z. J. Bie, H. Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2013, 52, 7451–7454 CrossRef CAS PubMed
- Z. Lin, L. X. Sun, W. Liu, Z. W. Xia, H. H. Yang and G. N. Chen, J. Mater. Chem. B, 2014, 2, 637–643 RSC
- J. Ye, Y. Chen and Z. Liu, Angew. Chem., Int. Ed., 2014, 53, 10386–10389 CrossRef CAS PubMed
- W. Zhang, W. Liu, P. Liu, H. B. Xiao, H. Wang and B. Tang, Angew. Chem., Int. Ed., 2014, 53, 12489–12493 CAS
- A. Nematollahzadeh, W. Sun, C. S. A. Aureliano, D. Lutkemeyer, J. Stute, M. J. Abdekhodaie, A. Shojaei and B. Sellergren, Angew. Chem., Int. Ed., 2011, 50, 495–498 CrossRef CAS PubMed
- J. X. Liu, K. G. Yang, Q. L. Deng, Q. R. Li, L. H. Zhang, Z. Liang and Y. K. Zhang, Chem. Commun., 2011, 47, 3969–3971 RSC
- W. H. Zhou, C. H. Lu, X. C. Guo, F. R. Chen, H. H. Yang and X. R. Wang, J. Mater. Chem., 2010, 20, 880–883 RSC
- R. X. Gao, X. R. Mu, Y. Hao, L. L. Zhang, J. J. Zhang and Y. H. Tang, J. Mater. Chem. B, 2014, 2, 1733–1741 RSC
- G. D. Brayer and A. McPherson, J. Biol. Chem., 1982, 257, 3359–3361 CAS
- G. Wohlfahrt, S. Witt, J. Hendle, D. Schomburg, H. M. Kalisz and H. J. Hecht, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1999, 55, 969–977 CrossRef CAS
- G. I. Berglund, G. H. Carlsson, A. T. Smith, H. Szoke, A. Henriksen and J. Hajdu, Nature, 2002, 417, 463–468 CrossRef CAS PubMed
- W. Zhou, N. Yao, G. P. Yao, C. H. Deng, X. M. Zhang and P. Y. Yang, Chem. Commun., 2008, 5577–5579 RSC
- Q. S. Li, F. Shen, X. Zhang, Y. F. Hu, Q. X. Zhang, L. Xu and X. Q. Ren, Anal. Chim. Acta, 2013, 795, 82–87 CrossRef CAS PubMed
- T. Takeuchi and T. Hishiya, Org. Biomol. Chem., 2008, 6, 2459–2467 CAS
- J. X. Liu, K. G. Yang, Y. Y. Qu, S. W. Li, Q. Wu, Z. Liang, L. H. Zhang and Y. K. Zhang, Chem. Commun., 2015, 51, 3896–3898 RSC
- Z. A. Lin, J. Wang, X. Q. Tan, L. X. Sun, R. F. Yu, H. H. Yang and G. N. Chen, J. Chromatogr. A, 2013, 1319, 141–147 CrossRef CAS PubMed
- S. H. Xuan, F. Wang, Y. Xiang, J. Wang, J. C. Yu and K. C. F. Leung, J. Mater. Chem., 2010, 20, 5086–5094 RSC
- S. H. Xuan, Y. Xiang, J. Wang, J. C. Yu and K. C. F. Leung, Chem. Mater., 2009, 21, 5079–5087 CrossRef CAS
- J. M. Guzman, S. A. Soper and R. L. McCarley, Anal. Chem., 2010, 82, 8970–8977 CrossRef PubMed
- K. Oshima, H. Toi and Y. Aoyama, Carbohydr. Lett., 1995, 1, 223–230 CAS
- S. Friedman, B. Pace and R. Pizer, J. Am. Chem. Soc., 1974, 96, 5381–5384 CrossRef CAS
- B. Yu, D. A. Wang, Q. Ye, F. Zhou and W. M. Liu, Chem. Commun., 2009, 6789–6791 RSC
- A. Postma, Y. Yan, Y. J. Wang, A. N. Zelikin, E. Tjipyo and F. Caruso, Chem. Mater., 2009, 21, 3042–3044 CrossRef CAS
- R. J. Umpleby, S. C. Baxter, Y. Z. Chen, R. N. Shah and K. D. Shimizu, Anal. Chem., 2011, 73, 4584–4591 CrossRef
Footnote |
| † Gang Wu and Jinyang Li contributed equally. |
| This journal is © The Royal Society of Chemistry 2015 |